Recently, an apprentice mentioned that she was reproducing the document: "automatic regulators for the continuation of erythroid differentiation reviewed by single cell transcript of human BM and UCB cells." When analyzing single cell data
She found that the author clearly mentioned six samples, but the data set was: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150774 , you can see five data:
GSM4558614 CD235a+ cells - umbilical cord blood 2 and umbilical cord blood 3,UCB3 GSM4558615 CD235a+ cells - umbilical cord blood 1 GSM4558616 CD235a+ cells - Bone Marrow 2 GSM4558617 CD235a+ cells - Bone Marrow 3 GSM4558618 CD235a+ cells - Bone Marrow 4
After carefully reading the description in the article, it is indeed the technology of 10x Genomics platform, which is 3 adult bone marrow and 3 umbilical cord blood samples. Together, there are 6 samples, and cell sorting is done in advance, just focusing on CD235a+ cells
The apprentice thought it was the author's data sorting and uploading failure. In fact, it was the cell hashing technology. You can first understand the cite SEQ technology, which can obtain the expression matrix of common genes and the expression matrix of dozens of proteins (through anti derived tags (ADT)) at the same time. The full name of this technology is cellular indexing of transcriptomes and Epitops by sequencing. Cell hashing is improved on the basis of cite SEQ. It is to add HTO (A distinct Hashtag oligonucleotide) labels to the samples that need to be mixed in advance, so that different HTO labels can be provided to distinguish even after mixing.
It can be seen that the file UCB2UCB3 is indeed mixed in the same single cell sample:
GSM4558614_UCB2UCB3_filtered_gene_bc_matrices.tar.gz 95.9 Mb GSM4558615_UCB1_filtered_gene_bc_matrices.tar.gz 4.4 Mb GSM4558616_BM2_filtered_gene_bc_matrices.tar.gz 7.4 Mb GSM4558617_BM3_filtered_gene_bc_matrices.tar.gz 6.3 Mb GSM4558618_BM4_filtered_gene_bc_matrices.tar.gz 8.3 Mb
Let's take a look at how to split this 95.9 Mb matrix into two samples
First import the data and view the data distribution
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
library(ggplot2)
# You need to go to the GSE150774 dataset home page to download GSE150774_RAW oh
# Take this GSM4558614_UCB2UCB3_filtered_gene_bc_matrices.tar.gz 95.9 Mb decompression:
HP <- Read10X("GSE150774_RAW/HP0_filtered_gene_bc_matrices/")
HP[[1]] # RNA: Gene Expression
HP[[2]] # HTO: Antibody Capture
In this folder, there are expression matrix and HTO (A distinct Hashtag oligonucleotide) tags, so that different HTO tags can be provided to distinguish even after mixing.
The expression matrix and HTO tag need to take the intersection
#Read hto data first yhto <- as.data.frame(HP$`Antibody Capture`) rownames(yhto) # You can see two samples table(colSums(yhto)== 0) pbmc.htos <- yhto[colSums(yhto)>0] dim(pbmc.htos) # Then look at the expression matrix pbmc.umis <- HP$`Gene Expression` # intersect joint.bcs <- intersect(colnames(pbmc.umis), colnames(pbmc.htos)) length(joint.bcs) # The previous antibody information and expression information need to be filtered # Subset RNA and HTO counts by joint cell barcodes pbmc.umis <- pbmc.umis[, joint.bcs] pbmc.htos <- as.matrix(pbmc.htos[, joint.bcs])
If your expression matrix and HTO label matrix are indeed the same experimental output, the intersection of the two should be very good.
Create Seurat and place HTO in object
After taking the intersection, you can carry out the standard process of seurat
# Setup Seurat object
pbmc.hashtag <- CreateSeuratObject(counts = pbmc.umis)
# Normalize RNA data with log normalization
pbmc.hashtag <- NormalizeData(pbmc.hashtag)
# Find and scale variable features
pbmc.hashtag <- FindVariableFeatures(pbmc.hashtag, selection.method = "mean.var.plot")
pbmc.hashtag <- ScaleData(pbmc.hashtag, features = VariableFeatures(pbmc.hashtag))
pbmc.hashtag
# Add HTO data as a new assay independent from RNA
pbmc.hashtag[["HTO"]] <- CreateAssayObject(counts = pbmc.htos)
names(pbmc.hashtag)
# Normalize HTO data, here we use centered log-ratio (CLR) transformation
pbmc.hashtag <- NormalizeData(pbmc.hashtag,
assay = "HTO",
normalization.method = "CLR")
head(pbmc.hashtag@meta.data)
VlnPlot(pbmc.hashtag, features = c("nFeature_RNA", "nCount_RNA",
"nCount_HTO","nFeature_HTO"),
ncol = 2,pt.size = 0)
You can see that at this time, both matrices are actually entered into the seurat object,
Splitting data using HTODemux function
The previous seurat object is special. It has two assessments, as shown below:
> pbmc.hashtag An object of class Seurat 32740 features across 18777 samples within 2 assays Active assay: RNA (32738 features, 1749 variable features) 1 other assay present: HTO
In this way, if there are two seurat objects of assembly, the data can be split by HTODemux function. The code is as follows:
pbmc.hashtag <- HTODemux(pbmc.hashtag,
assay = "HTO",
positive.quantile = 0.99)
pbmc.hashtag
table(pbmc.hashtag$HTO_classification.global)
pbmc.hashtag@assays
table(pbmc.hashtag$hash.ID)
# ## Doublet Negative Singlet
# 152 14650 3975
# visualization
Idents(pbmc.hashtag) <- "hash.ID"
VlnPlot(pbmc.hashtag, features = "nCount_RNA", pt.size = 0, log = TRUE)
FeatureScatter(pbmc.hashtag, feature1 = "rna_HBE1", feature2 = "rna_S100A9")
It can be seen that there is a problem with the quality of this data set. Most of the cells are negative, which is interesting.
Data extraction
Mix the samples and disassemble them into different seurat objects:
# First, we will remove negative cells from the object
table(Idents(pbmc.hashtag))
pbmc.hashtag.subset <- subset(pbmc.hashtag,
idents = c("Negative","Doublet"),
invert = TRUE)
table(Idents(pbmc.hashtag.subset))
pbmc.hashtag.subset <- RunPCA(pbmc.hashtag.subset)
pbmc.hashtag.subset <- RunUMAP(pbmc.hashtag.subset,dims=1:10)
DimPlot(pbmc.hashtag.subset)
#Extract B0251:
B0251 <- subset(pbmc.hashtag,
idents = c("B0251 anti-human Hashtag1"))
#Extract B0252:
B0252 <- subset(pbmc.hashtag,
idents = c("B0252 anti-human Hashtag2"))
Multiple single cell objects need to be merged and go through harmony process
sce.all <- merge( B0251, y= B0252 ,add.cell.ids = c('B0251','B0252'))
as.data.frame(sce.all@assays$RNA@counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all@meta.data$orig.ident)
library(stringr)
phe=as.data.frame(str_split(colnames(sce.all),'_',simplify = T))
head(phe)
sce.all@meta.data$orig.ident = phe$V1
table(sce.all@meta.data$orig.ident )
head(sce.all@meta.data)
# The following is the standard process
sce=sce.all
sce <- NormalizeData(sce,
normalization.method = "LogNormalize",
scale.factor = 1e4)
sce <- FindVariableFeatures(sce)
sce <- ScaleData(sce)
sce <- RunPCA(sce, features = VariableFeatures(object = sce))
library(harmony)
seuratObj <- RunHarmony(sce, "orig.ident")
names(seuratObj@reductions)
seuratObj <- RunUMAP(seuratObj, dims = 1:15,
reduction = "harmony")
DimPlot(seuratObj,reduction = "umap",label=T )
sce=seuratObj
sce <- FindNeighbors(sce, reduction = "harmony",
dims = 1:15)
sce.all=sce
The following results are logical. Group notes are as follows:
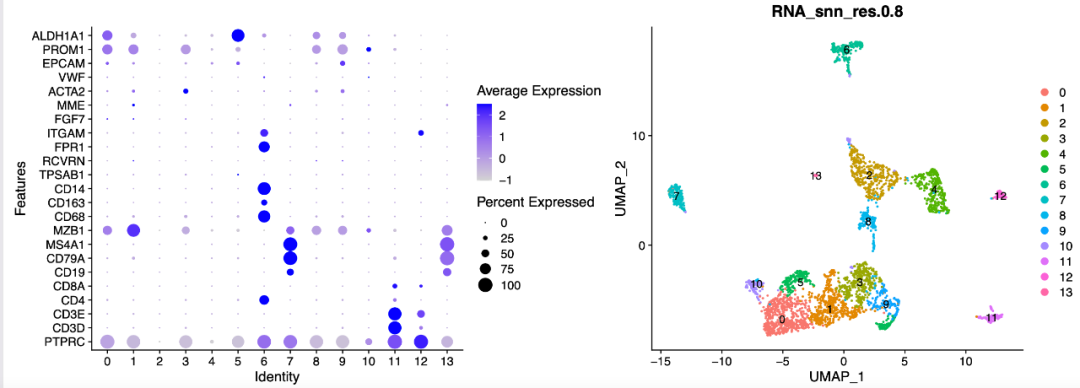
The only fly in the ointment is that the initial number of more than 20000 cells is only about 3000 after filtration.
If you really think my tutorial is helpful to your scientific research project and makes you enlightened, or your project uses a lot of my skills, please add a short thank you when publishing your achievements in the future, as shown below:
We thank Dr.Jianming Zeng(University of Macau), and all the members of his bioinformatics team, biotrainee, for generously sharing their experience and codes.
If I have traveled around the world in universities and research institutes (including Chinese mainland) in ten years, I will give priority to seeing you if you have such a friendship.