The review article "automatic cell type identification methods for single cell RNA sequencing", published in the Journal of computational and structural Biotechnology (October 2021), has sorted out the current annotation tools for single cell subsets. The link of the article is: https://www.sciencedirect.com/science/article/pii/S2001037021004499
- Lazy learning methods include CELLBLAST , scmap-cell , CellFishing.jl , and CellAtlasSearch .
- Eager learning methods account for the majority of the automatic methods, including scHPL , clustifyr , MARS , scPretrain , Superscan , Seurat , , scLearn , scCapsNet , ACTINN , CaSTLe , CHETAH , SciBet , scID , scmap-cluster , scPred , SingleCellNet , SingleR , scVI , scMatch , scClassifR , and Garnett .
- Marker learning methods include scTyper , DigitalCellSorter , SCINA , SCSA , CellAssign , and scCATCH . MarkerCount
- To facilitate automatic cell-type identification, scLearn, CELLBLAST, SciBet, SingleCellNet, scMatch, Superscan, and Garnett provide processed training datasets. Moreover, DigitalCellSorter, SCSA, scTyper, and scCATCH provide canonical cell markers for certain cell types.
The author has developed a package integrating so many tools (automatic cell type identification), which mainly divides each tool into three categories:
- eagersupervised methods include ACTINN, CaSTLe, CHETAH, clustifyr, Garnett, Markercount, MARS, scClassifR, scHPL, SciBet, scID, scLearn, scmapcluster, scPred, scVI, Seurat, SingleCellNet and SingleR.lazysupervised methods include CELLBLAST and scmapcell.markersupervised methods include scTyper, Markercount, SCSA, DigitalCellSorter and SCINA.
The workload is a little heavy!
However, the idea of software tool algorithm evaluation in the review article is worth learning:
- Fig. 1. Workflow of the traditional and automatic cell-type identification methods.
- Fig. 2. Performance of the automatic cell-type identification methods using the Tabula Muris datasets.
- Fig. 3. Performance of the automatic cell-type identification methods using PBMC and tumor datasets.
- Fig. 4. Speed of automatic cell-type identification methods.
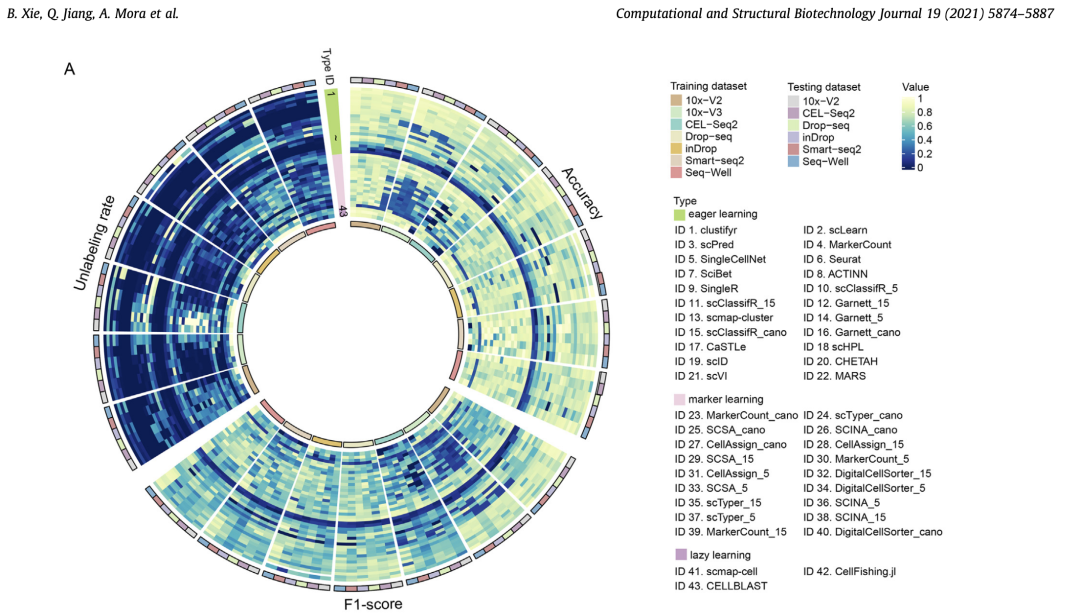
- Fig. 5. Summary of performance of the automatic cell-type identification methods. Bar graphs of the automatic cell-type identification methods with six evaluation criteria indicated.
The article also mentioned that at present, single-cell transcriptome sequencing data are multiple samples, so there are two problems to be solved:
- The first is to try to avoid the influences of different sequencing technologies during the process of data integration, for example, by using MNN , CCA , LIGER , Scanorama , et al.
- The second is to try to unify the currently inconsistent annotation levels in the training datasets, for example, by the joint usage of multiple training datasets , or by manual curation of each training dataset.
In fact, in a large number of tumor single cell data analysis projects I do, I don't use these automatic annotation tools. I see them with my naked eyes. I need some background knowledge! For example, recite the following list of genes with high expression in each cell subgroup:
# T Cells (CD3D, CD3E, CD8A), # B cells (CD19, CD79A, MS4A1 [CD20]), # Plasma cells (IGHG1, MZB1, SDC1, CD79A), # Monocytes and macrophages (CD68, CD163, CD14), # NK Cells (FGFBP2, FCG3RA, CX3CR1), # Photoreceptor cells (RCVRN), # Fibroblasts (FGF7, MME), # Endothelial cells (PECAM1, VWF). # epi or tumor (EPCAM, KRT19, PROM1, ALDH1A1, CD24). # immune (CD45+,PTPRC), epithelial/cancer (EpCAM+,EPCAM), # stromal (CD10+,MME,fibo or CD31+,PECAM1,endo)
Finally, the detailed GitHub web page links of various tools collected in this review article are excerpted:
Name of method Version URL CELLBLAST v0.3.8 https://github.com/gao-lab/Cell_BLAST CellFishing.jl v0.3.2 https://github.com/bicycle1885/CellFishing.jl scmap-cell v1.6.0 https://github.com/hemberg-lab/scmap ACTINN master https://github.com/mafeiyang/ACTINN CaSTLe v1.0.0.2 https://github.com/yuvallb/CaSTLe CHETAH v1.2.0 https://github.com/jdekanter/CHETAH Garnett v0.1.19 https://github.com/cole-trapnell-lab/garnett SciBet v0.1.0 https://github.com/zwj-tina/scibetR scID v2.1 https://github.com/BatadaLab/scID scLearn v1.0 https://github.com/bm2-lab/scLearn scmap-cluster v1.6.0 https://github.com/hemberg-lab/scmap scPred v1.9.0 https://github.com/powellgenomicslab/scPred scVI v0.4.1 https://github.com/YosefLab/scvi-tools Seurat v3.2.2 https://github.com/satijalab/seurat SingleCellNet v0.1.0 https://github.com/pcahan1/singleCellNet SingleR v1.1.1 https://github.com/dviraran/SingleR CellAssign v0.99.21 https://github.com/Irrationone/cellassign DigitalCellSorter v1.1 https://github.com/sdomanskyi/DigitalCellSorter SCINA v1.2.0 https://github.com/jcao89757/SCINA SCSA master https://github.com/bioinfo-ibms-pumc/SCSA scTyper v0.1.0 https://github.com/omicsCore/scTyper scHPL V0.0.2 https://github.com/lcmmichielsen/scHPL MARS master https://github.com/snap-stanford/mars clustifyr v1.5.0 https://github.com/rnabioco/clustifyr scClassifR v1.1.1 https://github.com/grisslab/scClassifR MarkerCount master https://github.com/combio-dku/MarkerCount/tree/master
Introduction to single-cell data processing requires some basic cognition. You can also see Lecture 10:
- 01. Upstream analysis process
- 02. How many samples of the subject and how much sequencing data
- 03. Filter unqualified cells and genes (data quality control is very important)
- 04. Filter mitochondrial ribosomal genes
- 05. Remove cellular and genetic effects
- 06. Dimensionality reduction clustering of single cell transcriptome data
- 07. Cell subpopulation annotation for single cell transcriptome data processing
- 08. Group the obtained subgroups more carefully
- 09. Comparison of cell subsets in single cell transcriptome data processing
The most basic is dimension reduction clustering. Refer to the previous example: Single cell clustering and clustering annotation that everyone can learn