Meet for the Winter Olympics
Main reference: Hail | GWAS Tutorial[1] this note aims to provide an overview of Hail functions, focusing on the functions of operating and querying genetic data sets. We conducted genome-wide SNP association tests and demonstrated the need to control confounding caused by population stratification.
1. Installation and environmental preparation
#Installing Jupyter
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
bash Miniconda3-latest-Linux-x86_64.sh
source miniconda3/bin/activate
conda create -n python36 python=3.6
conda activate python36
pip install hail==0.2.11
pip install jupyterlab
sudo apt-get install -y openjdk-8-jre-headless g++ libopenblas-base liblapack3
#Configure Jupyter
touch venv/conf.py
#Create password
PASSWD=$(python -c 'from notebook.auth import passwd; print(passwd("jup"))')
echo "c.NotebookApp.password = u'${PASSWD}'"
vi conf.py
c.NotebookApp.open_browser = False
c.NotebookApp.port = 8818
c.NotebookApp.password = u'sha1:{ sha1 value}' # Replace ${PASSWD} with the actual sha1 value
jupyter lab --config ./conf.py
# Create a new python notebook and initialize it
import hail as hl
hl.init()
# ############
using hail jar at /home/ubuntu/miniconda3/envs/python36/lib/python3.6/site-packages/hail/hail-all-spark.jar
Running on Apache Spark version 2.2.3
SparkUI available at http://10.0.8.13:4040
Welcome to
__ __ <>__
/ /_/ /__ __/ /
/ __ / _ `/ / /
/_/ /_/\_,_/_/_/ version 0.2.11-cf54f08305d1
LOGGING: writing to /home/ubuntu/hail/hail-20220203-1927-0.2.11-cf54f08305d1.log
According to the prompt, you can see the web page management interface of spark big data

Before using Hail, we imported some standard Python libraries for use in the whole notebook.
from hail.plot import show from pprint import pprint hl.plot.output_notebook()

This is a python drawing package similar to ggplot.
# Downloading 1kg data seems to be the website of Google. It was downloaded successfully
# We used a small portion of the common 1000 genome dataset, which was created by reducing the genotyping SNP in the complete VCF to about 20 MB sampling. We will also integrate sample and variant metadata from separate text files.
# These files are hosted by the Hail team in a public Google bucket; The following command downloads the data locally.
hl.utils.get_1kg('data/')
# ####################
2022-02-03 19:59:22 Hail: INFO: downloading 1KG VCF ...
Source: https://storage.googleapis.com/hail-tutorial/1kg.vcf.bgz
2022-02-03 19:59:24 Hail: INFO: importing VCF and writing to matrix table...
2022-02-03 19:59:29 Hail: INFO: Coerced sorted dataset
2022-02-03 19:59:40 Hail: INFO: wrote matrix table with 10879 rows and 284 columns in 16 partitions to data/1kg.mt
2022-02-03 19:59:40 Hail: INFO: downloading 1KG annotations ...
Source: https://storage.googleapis.com/hail-tutorial/1kg_annotations.txt
2022-02-03 19:59:40 Hail: INFO: downloading Ensembl gene annotations ...
Source: https://storage.googleapis.com/hail-tutorial/ensembl_gene_annotations.txt
2022-02-03 19:59:42 Hail: INFO: Done!
# View file status
!ls -lh data
# total 18M
-rw-r--r-- 1 ubuntu ubuntu 100K Feb 3 19:59 1kg_annotations.txt
drwxrwxr-x 7 ubuntu ubuntu 4.0K Feb 3 19:59 1kg.mt
-rw-r--r-- 1 ubuntu ubuntu 16M Feb 3 19:59 1kg.vcf.bgz
-rw-r--r-- 1 ubuntu ubuntu 2.5M Feb 3 19:59 ensembl_gene_annotations.txt
2. Practice your hands first and get familiar with the operation
The environment is well configured and you can study happily!
Import data from VCF [2]
The data in the VCF file is naturally represented as Hail MatrixTable[3]. By first importing the VCF file and then writing the generated MatrixTable in Hail's file format, all downstream operations on VCF data will be much faster.
hl.import_vcf('data/1kg.vcf.bgz').write('data/1kg.mt', overwrite=True)
# 2022-02-03 20:09:22 Hail: INFO: Coerced sorted dataset
2022-02-03 20:09:28 Hail: INFO: wrote matrix table with 10879 rows and 284 columns in 1 partition to data/1kg.mt
Next, we read the written file and allocate the variable MT (matrix table). mt = hl.read_matrix_table('data/1kg.mt')
Understand our data
It is important to have simple methods to slice, chunk, query and summarize data sets. Some of these functions are demonstrated below:
The rows[4] method can be used to obtain a table containing all row fields in MatrixTable.
We can use it with selection [5] to pull out five variants. This method uses a string or hail expression that refers to the field name in the table [6]. Here, we leave the parameter blank to keep only the row key and field. rows select locus alleles
mt.rows().select().show(5) # Or mt.row_key.show(5)

Use this method to display multiple attributes. show
mt.s.show(5)

To see the first few genotype calls, we can use entries[7] with select and take. The take method collects the first n rows into a list. Alternatively, we can use the show method to print the first n rows to the console in tabular format.
Try changing take to show in the following cell.
mt.entry.take(5) # ###### [Struct(GT=Call(alleles=[0, 0], phased=False), AD=[4, 0], DP=4, GQ=12, PL=[0, 12, 147]), Struct(GT=Call(alleles=[0, 0], phased=False), AD=[8, 0], DP=8, GQ=24, PL=[0, 24, 315]), Struct(GT=Call(alleles=[0, 0], phased=False), AD=[8, 0], DP=8, GQ=23, PL=[0, 23, 230]), Struct(GT=Call(alleles=[0, 0], phased=False), AD=[7, 0], DP=7, GQ=21, PL=[0, 21, 270]), Struct(GT=Call(alleles=[0, 0], phased=False), AD=[5, 0], DP=5, GQ=15, PL=[0, 15, 205])]
Change to show to make the table look better:

Add column field
Hail MatrixTable can have any number of row and column fields to store the data associated with each row and column. Annotation is usually a key part of any genetic study. The column fields are used to store information about the sample phenotype, ancestry, gender, and covariates. Row fields can be used to store information such as member genes and functional effects for use in QC or analysis.
In this tutorial, we will demonstrate how to get a text file and use it to annotate columns in MatrixTable.
The documents provided include sample ID, population (country) and "population (region)" name, sample gender and two simulated phenotypes (binary, or discrete).
This file can be imported_ Import table [8] into Hail. This function generates a Table[9] object. You can think of it as a Pandas or R data frame that is not limited by the memory on your computer - behind the scenes, it uses Spark.
# Import
table = (hl.import_table('data/1kg_annotations.txt', impute=True)
.key_by('Sample'))
# #############
2022-02-07 15:09:18 Hail: INFO: Reading table to impute column types
2022-02-07 15:09:18 Hail: INFO: Finished type imputation
Loading column 'Sample' as type 'str' (imputed)
Loading column 'Population' as type 'str' (imputed)
Loading column 'SuperPopulation' as type 'str' (imputed)
Loading column 'isFemale' as type 'bool' (imputed)
Loading column 'PurpleHair' as type 'bool' (imputed)
Loading column 'CaffeineConsumption' as type 'int32' (imputed)
A good way to look at a table's structure is to look at its schema.
table.describe()
# #############
----------------------------------------
Global fields:
None
----------------------------------------
Row fields:
'Sample': str
'Population': str
'SuperPopulation': str
'isFemale': bool
'PurpleHair': bool
'CaffeineConsumption': int32
----------------------------------------
Key: ['Sample']
----------------------------------------
To view the first few values, use the method show:
table.show(width=100) # 100 characters? # ########
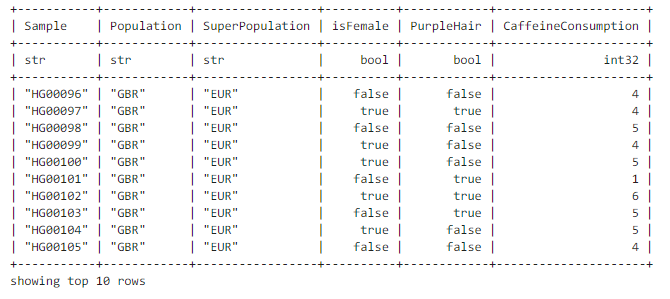
We will now use this table to add sample annotations to the dataset and store the annotations in the column fields of MatrixTable. First, we will print the existing column schema (similar to R language class?):
print(mt.col.dtype)
# struct{s: str}
We use annotate_ The cols [10] method joins the table with the MatrixTable containing the dataset.
mt = mt.annotate_cols(pheno = table[mt.s])
mt.col.describe()
# #############
--------------------------------------------------------
Type:
struct {
s: str,
pheno: struct {
Population: str,
SuperPopulation: str,
isFemale: bool,
PurpleHair: bool,
CaffeineConsumption: int32
}
}
--------------------------------------------------------
Source:
<hail.matrixtable.MatrixTable object at 0x7fa7f1a38278>
Index:
['column']
--------------------------------------------------------
Query function and Hail expression language
Hail has many useful query functions that can be used to collect statistics on data sets. These query functions take * * hail expression as parameter * *.
We will first look at some statistics of the information in the table. The aggregate[11] method can be used to aggregate rows in a table.
counter is an aggregate function that calculates the number of occurrences of each unique element. We can use it to see the distribution of population by passing Hail expression for the fields we want to count.
pprint(table.aggregate(hl.agg.counter(table.SuperPopulation)))
# ##########
{'AFR': 1018, 'AMR': 535, 'EAS': 617, 'EUR': 669, 'SAS': 661}
2022-02-07 15:22:59 Hail: INFO: Coerced sorted dataset
# It seems that the data set is not cleaned completely. Fortunately, it is just a warning and no error is reported.
stats is an aggregate function that generates useful statistics about a set of values. We can use it to observe the distribution of caffeine consumption phenotype.
pprint(table.aggregate(hl.agg.stats(table.CaffeineConsumption)))
# Several statistics
{'max': 10.0,
'mean': 3.983714285714278,
'min': -1.0,
'n': 3500,
'stdev': 1.7021055628070707,
However, these indicators do not fully represent the samples in our data set. The reasons are as follows:
table.count() # 3500 mt.count_cols() # 284 mt.count_cols() # 10879
Since the samples in the dataset are less than those in the complete thousand human genome project, we need to review the comments on the dataset. We can use aggregate_cols[12] to get only the indicators of the examples in the dataset.
mt.aggregate_cols(hl.agg.counter(mt.pheno.SuperPopulation))
# #########A lot of samples are missing
{'AFR': 76, 'EAS': 72, 'AMR': 34, 'SAS': 55, 'EUR': 47}
pprint(mt.aggregate_cols(hl.agg.stats(mt.pheno.CaffeineConsumption)))
{'max': 9.0,
'mean': 4.415492957746476,
'min': 0.0,
'n': 284,
'stdev': 1.5777634274659174,
'sum': 1253.9999999999993}
The functions demonstrated in the last few commands are not particularly new: it's certainly not difficult to solve these problems using Pandas or R data frames, or even Unix tools such as awk. But Hail can use the same interface and query language to analyze much larger sets, such as variation sets.
Here, we calculate the count of each SNP among the 12 possible unique SNPs (4 reference bases * 3 variant bases = 12 options).
To do this, we need to obtain the variant allele of each variant and then calculate the number of occurrences of each unique ref / alt pair. This can be done through Hail's counter function.
snp_counts = mt.aggregate_rows(hl.agg.counter(hl.Struct(ref=mt.alleles[0], alt=mt.alleles[1])))
pprint(snp_counts)
# ##########
{Struct(ref='A', alt='C'): 451,
Struct(ref='C', alt='A'): 494,
Struct(ref='C', alt='G'): 150,
Struct(ref='C', alt='T'): 2418,
Struct(ref='G', alt='A'): 2367,
Struct(ref='A', alt='T'): 75,
Struct(ref='T', alt='G'): 466,
Struct(ref='G', alt='C'): 111,
Struct(ref='G', alt='T'): 477,
Struct(ref='A', alt='G'): 1929,
Struct(ref='T', alt='A'): 77,
Struct(ref='T', alt='C'): 1864}
You can use Python's Counter class to list the counts in descending order.
from collections import Counter counts = Counter(snp_counts) counts.most_common() # ############ [(Struct(ref='C', alt='T'), 2418), (Struct(ref='G', alt='A'), 2367), (Struct(ref='A', alt='G'), 1929), (Struct(ref='T', alt='C'), 1864), (Struct(ref='C', alt='A'), 494), (Struct(ref='G', alt='T'), 477), (Struct(ref='T', alt='G'), 466), (Struct(ref='A', alt='C'), 451), (Struct(ref='C', alt='G'), 150), (Struct(ref='G', alt='C'), 111), (Struct(ref='T', alt='A'), 77), (Struct(ref='A', alt='T'), 75)]
It's nice to see that we can actually find something biological from this small data set: we see these frequencies appear in pairs. C/T and G / A are actually the same mutations, only observed from the opposite chain. Similarly, T/A and A/T are the same mutations on the opposite chain. There is a 30 fold difference between the frequencies of C/T and A/T SNP s. Why?
The same Python, R and Unix tools can do this, but we're starting to hit a wall - the latest version of gnomaD [13] has released about 250 million variants and can't be stored in memory on one computer.
What about the genotype? Hail can query the set of all genotypes in the dataset, which is getting larger and larger even for our small dataset. Our 284 samples and 10000 variants can produce 10 million unique genotypes. The gnomAD dataset has about 5 trillion unique genotypes.
The Hail drawing function allows the Hail field as a parameter, so we can directly pass the DP field here. If the range and bar column parameters are not set, this function calculates the range based on the minimum and maximum values of the field and uses the default of 50 columns.
p = hl.plot.histogram(mt.DP, range=(0,30), bins=30, title='DP Histogram', legend='DP') show(p)

The picture is still very beautiful
3. Quality control
Analysts spend most of their time on QC of sequenced data sets. QC is an iterative process, and each project is different: QC has no "button" solution. Every time Broad collects a new set of samples, it will find a new batch effect. However, by practicing open science and discussing QC processes and decisions with others, we can establish a set of best practices as a community.
QC is entirely based on the ability to understand dataset attributes. Hail tried to provide sample_qc[14] function to simplify this operation, which generates a set of useful indicators and stores them in the column field.
mt.col.describe()
# ##########
--------------------------------------------------------
Type:
struct {
s: str,
pheno: struct {
Population: str,
SuperPopulation: str,
isFemale: bool,
PurpleHair: bool,
CaffeineConsumption: int32
}
}
--------------------------------------------------------
Source:
<hail.matrixtable.MatrixTable object at 0x7fa7f1a38278>
Index:
['column']
--------------------------------------------------------
mt = hl.sample_qc(mt)
--------------------------------------------------------
Type:
struct {
s: str,
pheno: struct {
Population: str,
SuperPopulation: str,
isFemale: bool,
PurpleHair: bool,
CaffeineConsumption: int32
},
sample_qc: struct {
dp_stats: struct {
mean: float64,
stdev: float64,
min: float64,
max: float64
},
gq_stats: struct {
mean: float64,
stdev: float64,
min: float64,
max: float64
},
call_rate: float64,
n_called: int64,
n_not_called: int64,
n_hom_ref: int64,
n_het: int64,
n_hom_var: int64,
n_non_ref: int64,
n_singleton: int64,
n_snp: int64,
n_insertion: int64,
n_deletion: int64,
n_transition: int64,
n_transversion: int64,
n_star: int64,
r_ti_tv: float64,
r_het_hom_var: float64,
r_insertion_deletion: float64
}
}
mt.col.describe()
--------------------------------------------------------
Source:
<hail.matrixtable.MatrixTable object at 0x7fa7f09bd240>
Index:
['column']
--------------------------------------------------------
Drawing QC indicators is a good starting point.
p = hl.plot.histogram(mt.sample_qc.call_rate, range=(.88,1), legend='Call Rate') show(p)

Success rate of typing
p = hl.plot.histogram(mt.sample_qc.gq_stats.mean, range=(10,70), legend='Mean Sample GQ') show(p)
Usually, these indicators are relevant.
p = hl.plot.scatter(mt.sample_qc.dp_stats.mean, mt.sample_qc.call_rate, xlabel='Mean DP', ylabel='Call Rate') show(p)

Deleting outliers from the dataset will usually improve the correlation results. I don't know what DP and AD mean. Continue. We can make "any" cut-off and use them to filter:
mt = mt.filter_cols((mt.sample_qc.dp_stats.mean >= 4) & (mt.sample_qc.call_rate >= 0.97))
print('After filter, %d/284 samples remain.' % mt.count_cols())
# After filter, 250/284 samples remain.
Next is genotype QC. It is a good idea to filter out genotypes where the readings should not be: if we find a genotype called homozygous reference > 10% alt reads, or a genotype called heterozygous, without a ref / alt balance close to 1:1, it is likely to be a mistake.
In low depth data sets such as 1kg (PS.. Slightly changes cognition, 1kg is not highly sequenced, but the research standard of all humans), it is difficult to use this index to detect bad genotypes, because the reads ratio of 1 to 10 can be easily explained by binomial sampling. However, in the depth dataset, the reading ratio of 10:100 will certainly attract attention!
ab = mt.AD[1] / hl.sum(mt.AD)
filter_condition_ab = ((mt.GT.is_hom_ref() & (ab <= 0.1)) |
(mt.GT.is_het() & (ab >= 0.25) & (ab <= 0.75)) |
(mt.GT.is_hom_var() & (ab >= 0.9)))
fraction_filtered = mt.aggregate_entries(hl.agg.fraction(~filter_condition_ab))
print(f'Filtering {fraction_filtered * 100:.2f}% entries out of downstream analysis.')
mt = mt.filter_entries(filter_condition_ab)
# Filtering 3.63% entries out of downstream analysis.
Variation QC is the same: we can use variant_qc[15] function to generate various useful statistics, plot them and filter them.
mt = hl.variant_qc(mt)
mt.row.describe()
--------------------------------------------------------
Type:
struct {
locus: locus<GRCh37>,
alleles: array<str>,
rsid: str,
qual: float64,
filters: set<str>,
info: struct {
AC: array<int32>,
AF: array<float64>,
AN: int32,
BaseQRankSum: float64,
ClippingRankSum: float64,
DP: int32,
DS: bool,
FS: float64,
HaplotypeScore: float64,
InbreedingCoeff: float64,
MLEAC: array<int32>,
MLEAF: array<float64>,
MQ: float64,
MQ0: int32,
MQRankSum: float64,
QD: float64,
ReadPosRankSum: float64,
set: str
},
variant_qc: struct {
AC: array<int32>,
AF: array<float64>,
AN: int32,
homozygote_count: array<int32>,
dp_stats: struct {
mean: float64,
stdev: float64,
min: float64,
max: float64
},
gq_stats: struct {
mean: float64,
stdev: float64,
min: float64,
max: float64
},
n_called: int64,
n_not_called: int64,
call_rate: float32,
n_het: int64,
n_non_ref: int64,
het_freq_hwe: float64,
p_value_hwe: float64
}
}
--------------------------------------------------------
Source:
<hail.matrixtable.MatrixTable object at 0x7fa7f09dd6a0>
Index:
['row']
--------------------------------------------------------
These statistics actually look pretty good: we don't need to filter this dataset. However, most data sets require thoughtful quality control. filter_ The rows [16] method can help!
4. Start GWAS!
First, we need to limit to the following variants:
- Common (we will use the cut-off value of * * 1% *)
- Not far from the * * hardy Weinberg equilibrium * * [17], to find sequencing errors
mt = mt.filter_rows(mt.variant_qc.AF[1] > 0.01)
mt = mt.filter_rows(mt.variant_qc.p_value_hwe > 1e-6)
print('Samples: %d Variants: %d' % (mt.count_cols(), mt.count_rows()))
# Samples: 250 Variants: 7779
These filters removed about 15% of the sites (we started with more than 10000 sites). This does not mean that most sequencing data sets! We have downsampled a whole thousand genomic data sets to include more common variants than we accidentally expected.
In Hail, the association test accepts the column fields of the sample phenotype and covariates. We have obtained phenotypes of interest in the dataset (caffeine consumption):
gwas = hl.linear_regression_rows(y=mt.pheno.CaffeineConsumption,
x=mt.GT.n_alt_alleles(),
covariates=[1.0])
gwas.row.describe()
--------------------------------------------------------
Type:
struct {
locus: locus<GRCh37>,
alleles: array<str>,
n: int32,
sum_x: float64,
y_transpose_x: float64,
beta: float64,
standard_error: float64,
t_stat: float64,
p_value: float64
}
--------------------------------------------------------
Source:
<hail.table.Table object at 0x7fa7f0945828>
Index:
['row']
--------------------------------------------------------
Looking at the bottom of the printout above, you can see that linear regression adds new row fields for beta, standard error, t statistics, and p values.
Hail makes it easy to visualize the results! Let's make a Manhattan picture [18]:

It doesn't look like a skyline. Let's check whether our GWAS is well controlled using the Q-Q (quantile quantile) graph [19].
p = hl.plot.qq(gwas.p_value) show(p)

5. Confusion!
The observed p values will all deviate from expectations. There is either an unlikely causal relationship with each factor of caffeine intake (SNP).
We didn't tell you, but the sample ancestors were actually used to simulate this phenotype. This leads to * * stratified * * [20] distribution of phenotypes. The solution is to include ancestors as covariates in our regression.
linear_ regression_ The rows [21] function can also use column fields as covariates. We have annotated our samples with reported ancestors, but it is good to be skeptical about these labels due to human error. The genome does not have this problem! We will use the reported ancestors as genetic ancestors, but by including the calculated principal components in our model.
The pca[22] function generates the eigenvalues into a list, generates the example PC into a table, and can also generate variant loads when asked. hwe_ normalized_ The PCA [23] function does the same, using HWE normalized genotypes for PCA.
eigenvalues, pcs, _ = hl.hwe_normalized_pca(mt.GT) # ################## 2022-02-08 09:34:10 Hail: INFO: hwe_normalized_pca: running PCA using 7771 variants. 2022-02-08 09:34:15 Hail: INFO: pca: running PCA with 10 components... pprint(eigenvalues) # ########## [18.078002475659527, 9.978057054208994, 3.536048746504391, 2.6583611350017096, 1.5966283377451316, 1.5408505989074688, 1.5064316096906518, 1.4747771722760974, 1.4667919317402691, 1.4458363475579015] pcs.show(5, width=100)

Now that we have provided the principal components for each sample, we might as well draw them! Human history has had a strong impact on genetic data sets. Even with a 50 MB sequencing dataset, we can recover the main population.
mt = mt.annotate_cols(scores = pcs[mt.s].scores)
p = hl.plot.scatter(mt.scores[0],
mt.scores[1],
label=mt.pheno.SuperPopulation,
title='PCA', xlabel='PC1', ylabel='PC2')
show(p)

Now, we can rerun linear regression to control the sample gender and the first few principal components. We will do this as before, using the input variable instead of the number of alleles, and again using the genotype dose derived from the PL field by the input variable.
gwas = hl.linear_regression_rows(
y=mt.pheno.CaffeineConsumption,
x=mt.GT.n_alt_alleles(),
covariates=[1.0, mt.pheno.isFemale, mt.scores[0], mt.scores[1], mt.scores[2]])
# 2022-02-08 09:49:57 Hail: INFO: linear_regression_rows: running on 250 samples for 1 response variable y,
with input variable x, and 5 additional covariates...
We will first make a Q-Q diagram to evaluate the expansion

This shape indicates a well controlled (but not particularly effective) study. Now it's Manhattan:
p = hl.plot.manhattan(gwas.p_value) show(p)

We found a caffeine consumption point! Now just apply Hail's Nature paper function to publish the results.
Just kidding, this function won't log in until Hail 1.0!
Rare variation analysis
Here, we will demonstrate how to use the expression language to group and count any arbitrary attributes in row and column fields. Hail also implements the sequence core correlation test (SKAT).
entries = mt.entries()
results = (entries.group_by(pop = entries.pheno.SuperPopulation, chromosome = entries.locus.contig)
.aggregate(n_het = hl.agg.count_where(entries.GT.is_het())))
results.show()

We use matrixtable The entries [24] method converts the matrix table into a table (each sample of each variant has a row). In this representation, it is easy to aggregate any field we like, which is usually the first step in rare variant analysis.
What if we want to group by minor allele frequency and hair color and calculate the average GQ? We have shown that it is easy to aggregate through several arbitrary statistics. This particular example may not provide particularly useful information, but the same pattern can be used to detect the effects of rare variants:
entries = entries.annotate(maf_bin = hl.if_else(entries.info.AF[0]<0.01, "< 1%",
hl.if_else(entries.info.AF[0]<0.05, "1%-5%", ">5%")))
results2 = (entries.group_by(af_bin = entries.maf_bin, purple_hair = entries.pheno.PurpleHair)
.aggregate(mean_gq = hl.agg.stats(entries.GQ).mean,
mean_dp = hl.agg.stats(entries.DP).mean))
results2.show()
# module 'hail' has no attribute 'if_else'
# It's a little strange to report an error. The reason has not been found yet
The number of heterozygous genotypes of each gene was calculated according to the functional category (synonym, missense or loss of function) to estimate the functional constraints of each gene
The number of single loss of function mutations of each gene in the case was calculated and compared to detect the genes related to the disease.
epilogue
congratulations! You have reached the end of the first tutorial. To learn more about Hail's API and features, check out other tutorials. You can check the Python API[25] for documentation on other Hail functions. If you use Hail for your own science, we are happy to hear from you on Zulip chat [26] or forum [27].
As a reference, the following is the complete process of merging all tutorial endpoints into one script.
table = hl.import_table('data/1kg_annotations.txt', impute=True).key_by('Sample')
mt = hl.read_matrix_table('data/1kg.mt')
mt = mt.annotate_cols(pheno = table[mt.s])
mt = hl.sample_qc(mt)
mt = mt.filter_cols((mt.sample_qc.dp_stats.mean >= 4) & (mt.sample_qc.call_rate >= 0.97))
ab = mt.AD[1] / hl.sum(mt.AD)
filter_condition_ab = ((mt.GT.is_hom_ref() & (ab <= 0.1)) |
(mt.GT.is_het() & (ab >= 0.25) & (ab <= 0.75)) |
(mt.GT.is_hom_var() & (ab >= 0.9)))
mt = mt.filter_entries(filter_condition_ab)
mt = hl.variant_qc(mt)
mt = mt.filter_rows(mt.variant_qc.AF[1] > 0.01)
eigenvalues, pcs, _ = hl.hwe_normalized_pca(mt.GT)
mt = mt.annotate_cols(scores = pcs[mt.s].scores)
gwas = hl.linear_regression_rows(
y=mt.pheno.CaffeineConsumption,
x=mt.GT.n_alt_alleles(),
covariates=[1.0, mt.pheno.isFemale, mt.scores[0], mt.scores[1], mt.scores[2]])