Scanpy introduction and installation
Scanpy is an extensible toolkit for analyzing single cell analysis data constructed in conjunction with Ann data (a data structure).

For Windows systems, it is best to use the whl file for Scanpy installation. First download the whl file: scanpy-1.7.2-py3-none-any.whl
Through conda, use the command cd to enter the directory where the whl file is located, and then install through pip:
pip install scanpy-1.7.2-py3-none-any.whl
AnnData
Structure of AnnData
In scanpy, our most common data structure is AnnData, which is an object used to store data. Its data structure can be described as follows:
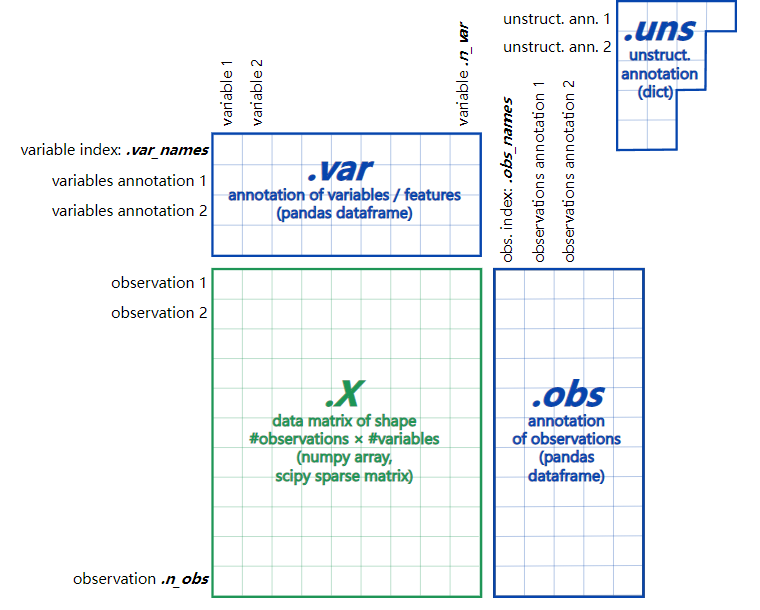
We record the above object as data. We need to understand the following parts:
| function | type | |
|---|---|---|
| adata.X | matrix | numpy matrix |
| adata.obs | Observation measurement | pandas Dataframe |
| adata.var | Characteristic quantity | pandas Dataframe |
| adata.uns | Unstructured data | Dictionary dict |
To further understand this data structure, we manually build an AnnData object:
import numpy as np import pandas as pd import anndata as ad from string import ascii_uppercase # Set the number of observation samples n_obs=1000 # obs is used to save observational information obs=pd.DataFrame() # numpy.random.choice(a, size=None, p=None) # Randomly extract elements from a(ndarray, but must be one-dimensional) and form an array of the specified size # Array p: corresponds to array a, indicating the probability of taking each element in array A. by default, the probability of selecting each element is the same obs['time']=np.random.choice(['day1','day2','day4','day8'],size=n_obs) # Set feature name var_names print(ascii_uppercase) # ABCDEFGHIJKLMNOPQRSTUVWXYZ var_names=[i*letter for i in range(1,10) for letter in ascii_uppercase] print(var_names) # ['A', 'B', 'C', 'D', 'E', 'F', 'G', 'H', 'I', ......, 'X', 'Y', 'Z', # ...... # 'AAAAAAAAA', 'BBBBBBBBB', 'CCCCCCCCC', ......, 'YYYYYYYYY', 'ZZZZZZZZZ'] # Number of features n_vars=len(var_names) # 234 # Defining a feature to a Dataframe var=pd.DataFrame(index=var_names) print(var.head()) # Now var has no columns (column index), only index (row index) # Create data matrix data. X X=np.arange(n_obs*n_vars).reshape(n_obs,n_vars)
Then initialize the andata object. The data type of the andata object is float32 by default. In order to facilitate later observation of the print results, we set the data type to int32:
adata=ad.AnnData(X,obs=obs,var=var,dtype='int32')
# View data
print(adata)
"""
AnnData object with n_obs × n_vars = 1000 × 234
obs: 'time'
"""
# View the X matrix of data
print(adata.X)
"""
[[ 0 1 2 ... 231 232 233]
[ 234 235 236 ... 465 466 467]
[ 468 469 470 ... 699 700 701]
...
[233298 233299 233300 ... 233529 233530 233531]
[233532 233533 233534 ... 233763 233764 233765]
[233766 233767 233768 ... 233997 233998 233999]]
"""
Generally, for data. X, rows correspond to observations (i.e., cells) and columns correspond to features (i.e., genes);
Each time we operate AnnData, instead of creating another AnnData to store data, we directly find the memory address of the previously initialized AnnData and directly change the value of AnnData through the memory address. The benefits are:
- No need to allocate extra memory;
- You can directly modify the initialized AnnoData object;
For example:
# View the first three elements of the 'A' column print(adata[:3, 'A'].X) """ [[ 0] [234] [468]] """ # Set the first three elements of the 'A' column adata[:3, 'A'].X = [0, 0, 0] # Looking at the first five elements of column 'A', we found that the value was modified print(adata[:5, 'A'].X) """ [[ 0] [ 0] [ 0] [702] [936]] """
However, if a part of the AnnData object is assigned to a new object, the object will get a new memory for storing the actual data instead of referring to the memory address of the original data object, such as:
adata_subset = adata[:5, ['A', 'B']]
print(adata_subset)
"""
View of AnnData object with n_obs × n_vars = 5 × 2
obs: 'time'
"""
# Create data for the new object_ Subset add observation 'foo'
adata_subset.obs['foo'] = range(5)
print(adata_subset)
"""
AnnData object with n_obs × n_vars = 5 × 2
obs: 'time', 'foo'
"""
h5ad: writing and reading of AnnData
We can save the AnnData object to disk through h5ad file. The saving process is as follows:
# Calculate the size of the object
def print_size_in_MB(x):
print('{:.3} MB'.format(x.__sizeof__()/1e6))
# View object size
print_size_in_MB(adata) # 0.187 MB
# Check whether to back up
print(adata.isbacked) # False
# Set backup address
adata.filename = './test.h5ad'
# Check whether the backup was successful
print(adata.isbacked) # True
When the status of data.isbacked is True, the object has been written to the disk;
On the contrary, we can use scanpy to easily read the file and obtain the AnnData object:
import scanpy as sc
Myadata=sc.read('./test.h5ad')
print(Myadata)
"""
AnnData object with n_obs × n_vars = 1000 × 234
obs: 'time'
"""
Usage of some common APIs in Scanpy
Import Scanpy first:
import scanpy as sc
sc.pp.filter_cells
sc.pp.filter_cells(data, min_genes=None, max_genes=None)
It is often used in pretreatment to do some cell screening, and the function is retained for at least min_genes are cells with more than one gene, or retain at most max_genes: cells with multiple genes;
Also note that the parameter min_genes and parameter max_genes cannot be passed at the same time;
example:
# Import data
adata=sc.datasets.krumsiek11() # Five types of cells, 640 cell samples, and a total of 11 genes were measured
print(adata)
"""
AnnData object with n_obs × n_vars = 640 × 11
obs: 'cell_type'
uns: 'iroot', 'highlights'
"""
print(adata.n_obs) # 640 cells
# 11 genes (i.e. characteristics)
print(adata.var_names)
"""
Index(['Gata2', 'Gata1', 'Fog1', 'EKLF', 'Fli1', 'SCL', 'Cebpa', 'Pu.1',
'cJun', 'EgrNab', 'Gfi1'],
dtype='object')
"""
### Observe the change of cell number ###
sc.pp.filter_cells(adata,min_genes=0) # Equivalent to no filtering
print(adata.n_obs) # 640
print(adata.obs)
"""
cell_type n_genes
0 progenitor 9
.. ... ...
159 Neu 8
"""
print(set(adata.obs['cell_type'].values)) # Class 5 cells {'Neu', 'progenitor', 'Ery', 'Mo', 'Mk'}
print(adata.obs['n_genes'].min()) # 4. At least four genes were measured in each cell
sc.pp.filter_cells(adata,min_genes=6) # Cells with more than 6 genes were selected and measured
print(adata.n_obs) # 630
print(adata.obs['n_genes'].min()) # 6
sc.pp.filter_genes
sc.pp.filter_genes(data, min_cells=None, max_cells=None)
This function is used to keep at least min_cells are genes that appear in more than one cell or remain at most_ Cells are genes that appear in cells;
Parameter min_cells and parameter max_cells cannot be passed at the same time;
Compare sc.pp.filter_cells can be found, sc.pp.filter_genes is used to select genes (screening column), sc.pp.filter_cells are used to select cells (filter rows);
sc.pp.highly_variable_genes
sc.pp.highly_variable_genes( data, n_top_genes=None, min_disp=0.5, max_disp=inf, min_mean=0.0125, max_mean=3)
This function is used to determine hypervariable genes;
Description of common parameters:
- data: AnnData Matrix, the row corresponds to the cell, and the column corresponds to the gene
- n_top_genes: the number of hypervariable genes to retain
sc.pp.normalize_total
sc.pp.normalize_total(adata, target_sum=None, inplace=True)
The function can standardize each cell so that each cell can sum along the gene direction after standardization and have the same total target_sum;
example:
adata.X
array([[ 3., 3., 3., 6., 6.],
[ 1., 1., 1., 2., 2.],
[ 1., 22., 1., 2., 2.]], dtype=float32)
# Set target_sum=1 after standardization
X_norm
array([[0.14, 0.14, 0.14, 0.29, 0.29],
[0.14, 0.14, 0.14, 0.29, 0.29],
[0.04, 0.79, 0.04, 0.07, 0.07]], dtype=float32)