The double hybrid functional was first proposed by Grimme in 2006. Its basic idea is to introduce the information of unoccupied orbits into the exchange correlation functional in the form of second-order perturbation. It belongs to the fifth-order functional on the Jacob ladder of density functional, which has high accuracy. Double hybrid functional is a good choice when CCSD(T) cannot be used due to the high requirement of energy accuracy. Since the energy in the form of MP2 is introduced, its calculation scale is O(N5). Most common quantum chemical programs support double hybrid functional, and ORCA is very efficient because it supports RI and DLPNO approximation. This paper mainly introduces the use of double hybrid functional in ORCA. The version used is ORCA 5.0.2.
1, Single point energy calculation
Taking the structure of A-T base pair connected by hydrogen bond as an example, the structure is as follows:
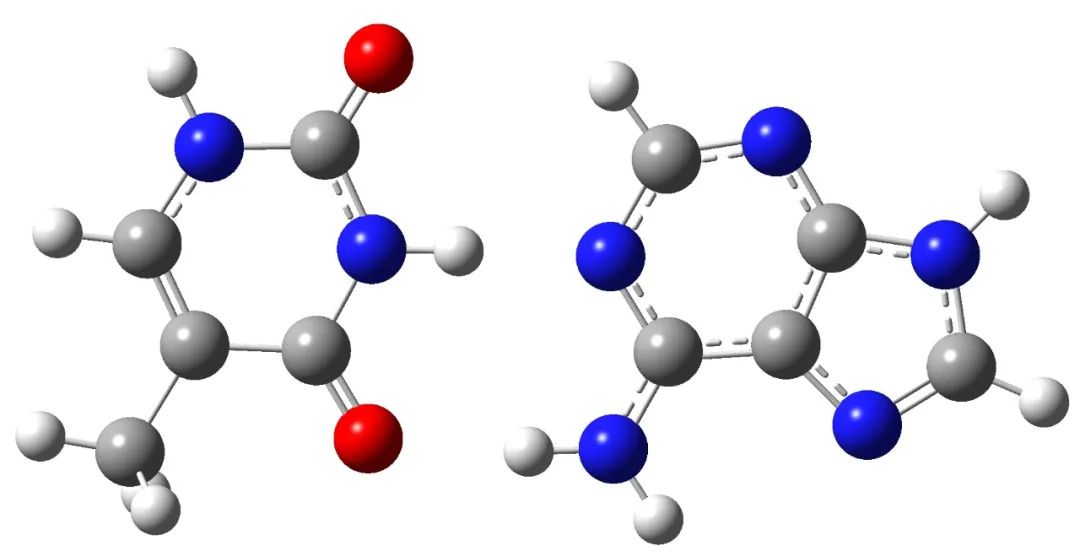
The input file is as follows:
%pal nprocs 52 end %maxcore 2000 ! PWPB95 def2-TZVP def2-TZVP/C tightSCF *xyz 0 1 N 0.9350155 -0.0279801 -0.3788916 C 1.6739638 -0.0357766 0.7424316 C 3.0747955 -0.0094480 0.5994562 C 3.5646109 0.0195446 -0.7059872 N 2.8531510 0.0258031 -1.8409596 C 1.5490760 0.0012569 -1.5808009 N 4.0885824 -0.0054429 1.5289786 C 5.1829921 0.0253971 0.7872176 N 4.9294871 0.0412404 -0.5567274 N 1.0716177 -0.0765366 1.9391390 H 0.8794435 0.0050260 -2.4315709 H 6.1882591 0.0375542 1.1738824 H 5.6035368 0.0648755 -1.3036811 H 0.0586915 -0.0423765 2.0039181 H 1.6443796 -0.0347395 2.7619159 N -3.9211729 -0.0009646 -1.5163659 C -4.6136833 0.0169051 -0.3336520 C -3.9917387 0.0219348 0.8663338 C -2.5361367 0.0074651 0.8766724 N -1.9256484 -0.0110593 -0.3638948 C -2.5395897 -0.0149474 -1.5962357 C -4.7106131 0.0413373 2.1738637 O -1.8674730 0.0112093 1.9120833 O -1.9416783 -0.0291878 -2.6573783 H -4.4017172 -0.0036078 -2.4004924 H -0.8838255 -0.0216168 -0.3784269 H -5.6909220 0.0269347 -0.4227183 H -4.4439282 -0.8302573 2.7695655 H -4.4267056 0.9186178 2.7530256 H -5.7883971 0.0505530 2.0247280 *
Starting from ORCA 5.0, RIJCOSX approximation is turned on by default when calculating hybrid and double hybrid functional (SCF part of double hybrid), while RI approximation is turned on by default when calculating relevant energy part. For RIJCOSX approximation, there is no need to specify the auxiliary base, and the program will automatically select it; For the calculation of related energy, you need to manually specify the auxiliary basis, such as def2-TZVP/C in this example. In addition, for double hybrid functional, D3 correction can also be added, just write D3 on the keyword line. For super large systems, DLPNO approximation is also supported. The keyword is written by adding DLPNO prefix to the functional, such as DLPNO-PWPB95.
We do not show the specific output file here. The result of single point energy can be seen by looking for "FINAL SINGLE POINT ENERGY" in the output file.
2, Geometry optimization
ORCA supports the analytical gradient of double hybrid functional, and it also supports DLPNO version. Therefore, double hybrid functional can be used for geometric structure optimization. In ORCA, many double hybrid functionals do not support analytical gradients, such as the above PWPB95 functionals. The frequency calculation of double hybrid functional analysis is not supported in ORCA 5.0.2, so it is impossible to use opt freq combination to calculate the frequency after optimizing the structure. Incidentally, in ORCA, standard MP2 and RI-MP2 support analytical two-step degree, but do not support frozen core approximation. Therefore, if frequency calculation is to be done, it needs to be closed Frozen core approximation , use the keyword nofrozencore.
Here, we take the stacked A-T complex as an example to optimize the geometric structure. Its structure is as follows:
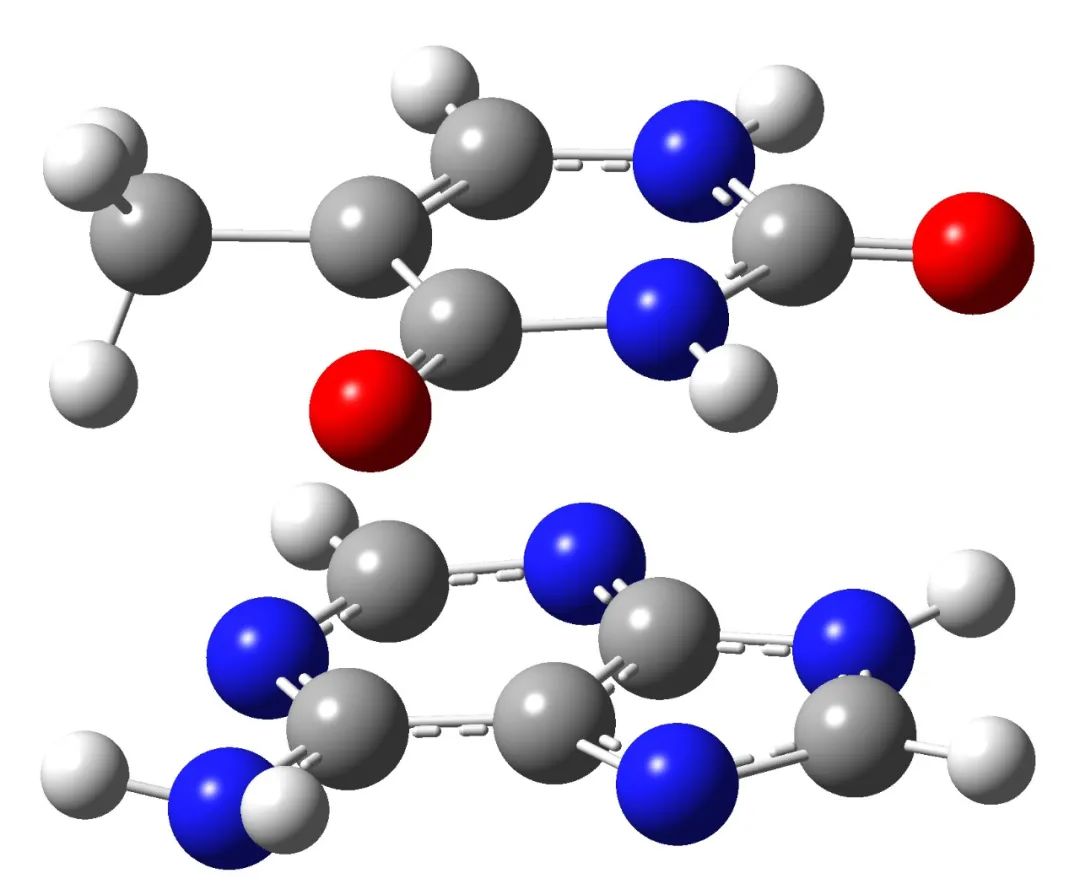
The input file is as follows:
%maxcore 10000 %pal nprocs 52 end ! opt wB97X-2 def2-SVP def2-SVP/C *xyz 0 1 N 0.2793014 2.4068393 -0.6057517 C -1.0848570 2.4457461 -0.5511608 H -1.6594403 3.0230294 -1.2560905 N -1.5977117 1.7179877 0.4287543 C -0.4897255 1.1714358 1.0301910 C -0.3461366 0.2914710 2.1172343 N -1.4187090 -0.1677767 2.8101441 H -1.2388750 -0.9594802 3.4047578 H -2.2918734 -0.1788223 2.3073619 N 0.8857630 -0.0700763 2.4919494 C 1.9352348 0.4072878 1.7968022 H 2.9060330 0.0788414 2.1458181 N 1.9409775 1.2242019 0.7402202 C 0.6952186 1.5779858 0.4063984 H 0.8610073 2.8298045 -1.3104502 N 1.2754606 -0.6478993 -1.9779104 C 1.4130533 -1.5536850 -0.9550667 H 2.4258769 -1.8670780 -0.7468778 C 0.3575976 -2.0239499 -0.2530575 C 0.4821292 -3.0179494 0.8521221 H 0.1757705 -2.5756065 1.7986281 H -0.1601691 -3.8770412 0.6639498 H 1.5112443 -3.3572767 0.9513659 C -0.9684711 -1.5298112 -0.5939792 O -2.0029280 -1.8396957 -0.0199453 N -0.9956916 -0.6383870 -1.6720420 H -1.9014057 -0.2501720 -1.8985760 C 0.0684702 -0.1191762 -2.3763759 O -0.0397875 0.7227006 -3.2531083 H 2.0853289 -0.2760176 -2.4454577 *
For the interpretation of ORCA geometric structure optimization results, please refer to< Using ORCA for structural optimization and track viewing >A penny.
3, Excited state calculation
The double hybrid functional also supports the calculation of the properties of excited states in the TD-DFT framework, and more double hybrid functional optimized for excited states are supported in ORCA, such as ω B2PLYP. Taking pyridine molecule as an example, the input file is as follows:
%maxcore 4000 %pal nprocs 40 end ! wB2GP-PLYP def2-TZVP def2-TZVP/C %tddft nroots 10 end *xyz 0 1 C -1.29882942 0.07547631 0.00000000 C 0.10804858 0.07547631 0.00000000 C 0.77473958 1.30208331 0.00000000 C 0.02066658 2.47687031 -0.00024100 C -1.38253542 2.37406531 -0.00038200 N -2.04206542 1.19911431 -0.00019700 H 1.87328358 1.34185931 -0.00095000 H -1.86437642 -0.87334469 0.00012200 H 0.66278058 -0.87142469 0.00026400 H 0.50470258 3.46179431 -0.00034200 H -2.01588442 3.27912831 -0.00046300 *
The result output is similar to that of ordinary functional, as follows:
-----------------------------------------------------------------------------
ABSORPTION SPECTRUM VIA TRANSITION ELECTRIC DIPOLE MOMENTS
-----------------------------------------------------------------------------
State Energy Wavelength fosc T2 TX TY TZ
(cm-1) (nm) (au**2) (au) (au) (au)
-----------------------------------------------------------------------------
1 42738.1 234.0 0.005922762 0.04562 -0.00011 -0.00019 -0.21360
2 44845.5 223.0 0.056651237 0.41588 0.02318 -0.64447 0.00014
3 53724.7 186.1 0.024913935 0.15267 0.39050 0.01315 -0.00011
4 45605.6 219.3 0.000000221 0.00000 -0.00089 -0.00089 0.00007
5 63005.4 158.7 0.826929088 4.32082 -0.08234 2.07703 -0.00024
6 63741.9 156.9 0.875186430 4.52013 -2.12439 -0.08427 -0.00087
7 66806.1 149.7 0.000001593 0.00001 0.00011 0.00279 0.00018
8 66974.5 149.3 0.000000053 0.00000 0.00016 -0.00004 -0.00049
9 71441.4 140.0 0.018016928 0.08302 -0.00073 -0.00006 -0.28814
10 70282.5 142.3 0.009596157 0.04495 -0.00027 -0.00003 0.21201Summary
This paper introduces how to use double hybrid functional to calculate single point energy, geometric structure optimization and excited states in ORCA. In addition to these three types of calculations, double hybrid functional can also be used in ORCA for other types of calculations, such as NMR properties. For details, please refer to the manual.