Everything is difficult at the beginning. Considering that many small partners often read the expression matrix uploaded by the author in the first step to build the seurat object when doing single-cell public data analysis, it is very necessary to sort out the 18 single-cell data format files.
Now let's demonstrate how to read a single cell file in room format. First, install and load some packages:
library(hdf5r)
library(loomR)
library(LoomExperiment)
# remotes::install_github("aertslab/SCopeLoomR")
# remotes::install_local('mojaveazure-loomR-0.2.0-beta-54-g1eca16a.tar.gz')
library(SCopeLoomR)
It is worth noting that some packages are actually on GitHub. If your network is poor, you need to find a solution by yourself. If you can't install package reading, you might as well try our * * marathon teaching (live one month interactive teaching), and you can watch the 200 Q & a selected from more than 2000 questions and interactive exchanges! 2021 phase II_ Introduction class of Shengxin_ Wechat group Q & a sorting , and 2021 phase II_ Data mining class_ Wechat group Q & A notes
With 100000 students, you deserve the following classes:
- Data mining (GEO,TCGA, single cell) 2021 issue 11 (annual closing)
- Introduction to student information-2021 issue 11 (annual closing)
Next, we take gse160756 as an example to enter https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE160756 It can be seen that the seven samples of its data set are shared with you in room format.
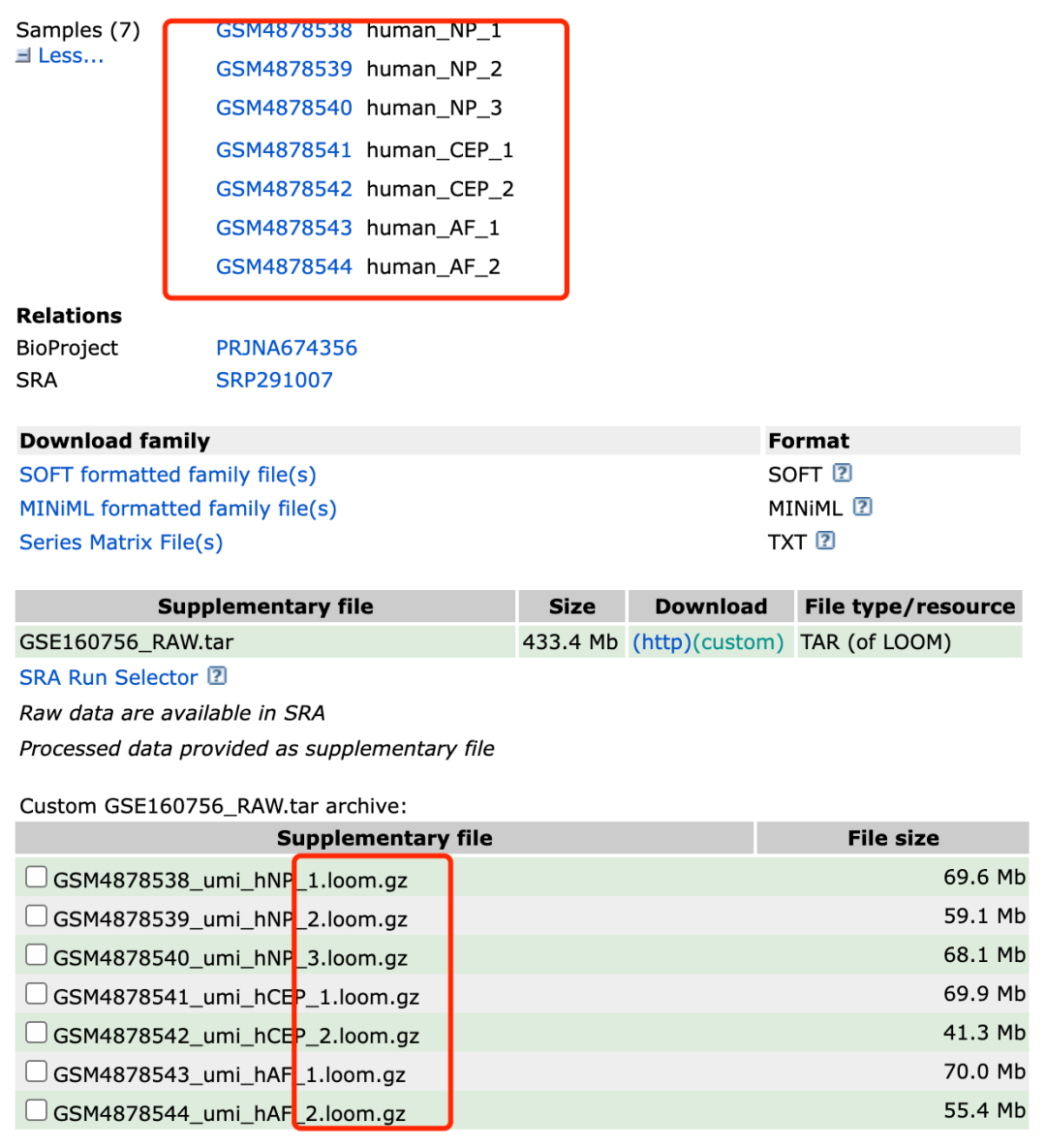
Share it with you in loom format
Our example code is as follows;
###### step1:Import data ######
path='GSE160756_RAW/'
samples=list.files(path )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
folder=file.path(path,pro)
print(pro)
print(folder)
# Import room file
lfile <- connect(filename =folder, mode = "r+")
ct <- as.data.frame(lfile[["matrix"]][,])
colnames(ct) <- lfile[["row_attrs/gene_names"]][] # Add gene_name
rownames(ct) <- lfile[["col_attrs/cell_names"]][] # Add cell_name
ct[1:5,1:5]
ct <- t(ct) # Note transpose
sce=CreateSeuratObject(counts = ct,
project = pro )
return(sce)
})
names(sceList)
samples
After reading all the room files, it becomes a list. There are independent seurat objects for each sample, which need to be merged. The code is as follows:
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples)
as.data.frame(sce.all@assays$RNA@counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all@meta.data$orig.ident)
library(stringr)
phe=str_split(rownames(sce.all@meta.data),'_',simplify = T)
head(phe)
table(phe[,4])
table(phe[,3])
sce.all@meta.data$rep=phe[,4]
sce.all@meta.data$group=phe[,3]
sce.all@meta.data$orig.ident = paste0(
sce.all@meta.data$group,
sce.all@meta.data$rep
)
table(sce.all@meta.data$orig.ident)
Because there are multiple samples, it is usually recommended to follow the harmnoy process for integration.
You can find 10x single cell transcriptome data in the following articles, which are published in the link of geo database:
- My subject has only one 10x sample. What should I do?
- 10x single cell transcriptome data analysis strategy for two samples
- CCA integration of three 10X single cell transcriptome samples
- CCA Seurat package for data integration of multiple single cell transcriptome samples
If you don't have a basic understanding of single cell data analysis, you can see basic 10:
- 01. Upstream analysis process
- 02. How many samples of the subject and how much sequencing data
- 03. Filter unqualified cells and genes (data quality control is very important)
- 04. Filter mitochondrial ribosomal genes
- 05. Remove cellular and genetic effects
- 06. Dimensionality reduction clustering of single cell transcriptome data
- 07. Cell subpopulation annotation for single cell transcriptome data processing
- 08. Group the obtained subgroups more carefully
- 09. Comparison of cell subsets in single cell transcriptome data processing
The most basic is dimension reduction clustering. Refer to the previous example: Single cell clustering and clustering annotation that everyone can learn