Mike L. Smith, Steffen Durinck, Wolfgang Huber
12 August 2021
Package: biomaRt 2.49.4
introduction
By far, the biomaRt package is the most commonly used to access the data available in Ensembl. With this in mind, biomaRt provides many features customized specifically for the biomaRt instance provided by Ensembl. This article details this Ensembl specific functionality and provides a number of sample use cases that can be used as a basis for specifying your own queries.
Select the Ensembl BioMart database and dataset
Each analysis using biomaRt starts with selecting the biomaRt database to use. The following command will connect us to the latest version of Human Genes BioMart of Ensembl.
> library(biomaRt) > ensembl <- useEnsembl(biomart = "genes", dataset = "hsapiens_gene_ensembl")
If this is your first time using biomaRt, you may want to know how to find the two parameters we provided to the useEnsembl() command. This is a two-step process, but once you know the required settings, you can use the version shown above as a single command. These initial steps are outlined below.
Step 1: determine the database you need
The first step is to find the name of the biomaRt service currently provided by Ensembl. We can do this using the function listEnsembl(), which will display all available Ensembl BioMart Web services. The first column gives the name we should provide for the biomaRt parameter in useEnsembl(), and the second column gives a more comprehensive title of the dataset and the Ensembl version.
> listEnsembl()
biomart version
1 genes Ensembl Genes 104
2 mouse_strains Mouse strains 104
3 snps Ensembl Variation 104
4 regulation Ensembl Regulation 104
You can now connect to the required biomaRt database using the useEnsembl() function. You should provide a valid name for the biomaRt parameter from the output of listEnsembl(). In the next example, we will select the main Ensembl mart, which provides access to gene annotation information.
> ensembl <- useEnsembl(biomart = "genes")
If we print the current ensembl object, we can see ENSEMBL_MART_ENSEMBL database 1 is selected, but no dataset is selected.
> ensembl Object of class 'Mart': Using the ENSEMBL_MART_ENSEMBL BioMart database No dataset selected.
Step 2: select a dataset
The BioMart database can contain multiple data sets. For example, in the Ensembl gene mart, each species is a different dataset. In the next step, we use the function listDatasets() to view the datasets available in the selected BioMart.
Note: Here we use the function head() to display only the first five entries, because the complete list has 202 entries.
> datasets <- listDatasets(ensembl)
> head(datasets)
dataset description version
1 abrachyrhynchus_gene_ensembl Pink-footed goose genes (ASM259213v1) ASM259213v1
2 acalliptera_gene_ensembl Eastern happy genes (fAstCal1.2) fAstCal1.2
3 acarolinensis_gene_ensembl Green anole genes (AnoCar2.0v2) AnoCar2.0v2
4 acchrysaetos_gene_ensembl Golden eagle genes (bAquChr1.2) bAquChr1.2
5 acitrinellus_gene_ensembl Midas cichlid genes (Midas_v5) Midas_v5
6 amelanoleuca_gene_ensembl Giant panda genes (ASM200744v2) ASM200744v2
The listDatasets() function returns every available option, but when the list of results is long, it can be cumbersome and requires a lot of scrolling to find the item you are interested in. biomaRt also provides the searchDatasets() function, which attempts to find any entries that match a particular term or pattern. For example, if we want to find details of any dataset containing the term "hsapiens" in our ensembl mart, we can do the following:
> searchDatasets(mart = ensembl, pattern = "hsapiens")
dataset description version
80 hsapiens_gene_ensembl Human genes (GRCh38.p13) GRCh38.p13
To use the dataset, we can update our Mart object using the function useDataset(). In the following example, we chose to use the hsapiens dataset.
> ensembl <- useDataset(dataset = "hsapiens_gene_ensembl", mart = ensembl)
As mentioned earlier, if you know the dataset to be used in advance, that is, you have completed this process before, we can select the database and dataset in one step:
> ensembl <- useEnsembl(biomart = "genes", dataset = "hsapiens_gene_ensembl")
Ensembl mirror site
To improve performance, Ensembl provides multiple mirrors of sites distributed around the world. When you use the default setting of useEnsembl(), your query will be directed to the image closest to the geographic location. In theory, this should give you the best performance, but not always in practice. For example, if a recent mirror encounters many queries from other users, it may not perform well for you. You can explicitly request a specific mirror using the mirror parameter useEnsembl().
> ensembl <- useEnsembl(biomart = "ensembl",dataset = "hsapiens_gene_ensembl",mirror = "useast")
The values of the mirror parameter are: us Eastern, us west, asia, and www.
Use the archived version of Ensembl
The archived version of Ensembl can be queried through biomaRt, so you can maintain consistent comments throughout the project.
biomaRt provides the function listEnsemblArchives() to view available Ensembl files. This function takes no arguments and generates a table containing the names and version numbers of available archives, the first release date, and the URL s that can access them.
> listEnsemblArchives()
name date url version current_release
1 Ensembl GRCh37 Feb 2014 https://grch37.ensembl.org GRCh37
2 Ensembl 104 May 2021 https://may2021.archive.ensembl.org 104 *
3 Ensembl 103 Feb 2021 https://feb2021.archive.ensembl.org 103
4 Ensembl 102 Nov 2020 https://nov2020.archive.ensembl.org 102
5 Ensembl 101 Aug 2020 https://aug2020.archive.ensembl.org 101
6 Ensembl 100 Apr 2020 https://apr2020.archive.ensembl.org 100
7 Ensembl 99 Jan 2020 https://jan2020.archive.ensembl.org 99
8 Ensembl 98 Sep 2019 https://sep2019.archive.ensembl.org 98
9 Ensembl 97 Jul 2019 https://jul2019.archive.ensembl.org 97
10 Ensembl 96 Apr 2019 https://apr2019.archive.ensembl.org 96
11 Ensembl 95 Jan 2019 https://jan2019.archive.ensembl.org 95
12 Ensembl 94 Oct 2018 https://oct2018.archive.ensembl.org 94
13 Ensembl 93 Jul 2018 https://jul2018.archive.ensembl.org 93
14 Ensembl 92 Apr 2018 https://apr2018.archive.ensembl.org 92
15 Ensembl 91 Dec 2017 https://dec2017.archive.ensembl.org 91
16 Ensembl 90 Aug 2017 https://aug2017.archive.ensembl.org 90
17 Ensembl 89 May 2017 https://may2017.archive.ensembl.org 89
18 Ensembl 88 Mar 2017 https://mar2017.archive.ensembl.org 88
19 Ensembl 87 Dec 2016 https://dec2016.archive.ensembl.org 87
20 Ensembl 86 Oct 2016 https://oct2016.archive.ensembl.org 86
21 Ensembl 85 Jul 2016 https://jul2016.archive.ensembl.org 85
22 Ensembl 80 May 2015 https://may2015.archive.ensembl.org 80
23 Ensembl 77 Oct 2014 https://oct2014.archive.ensembl.org 77
24 Ensembl 75 Feb 2014 https://feb2014.archive.ensembl.org 75
25 Ensembl 67 May 2012 https://may2012.archive.ensembl.org 67
26 Ensembl 54 May 2009 https://may2009.archive.ensembl.org 54
Alternatively, you can use http://www.ensembl.org Find archived versions on the web site. Scroll down from the home page to the bottom of the page, click View in archive and select the archive you want.
You will notice that even the current version of Ensembl has an archive URL. It is useful if you want to ensure that the script you write now returns exactly the same results in the future. Using www.ensembl.org will always access the current version, so the retrieved data may change with the emergence of a new version.
Whichever method you use to find the url of the archive to query, copy the url and use it in the host parameters to connect to the specified BioMart database as shown below. The following example shows how to query Ensembl 54.
> listEnsembl(version = 95)
biomart version
1 genes Ensembl Genes 95
2 mouse_strains Mouse strains 95
3 snps Ensembl Variation 95
4 regulation Ensembl Regulation 95
> ensembl95 <- useEnsembl(biomart = 'genes',dataset = 'hsapiens_gene_ensembl',version = 95)
Using Ensembl Genomes
Ensembl Genomes has expanded its scope of work by providing annotations from the vertebrate genomes provided by the main Ensembl project, crossing the classification space and providing a separate BioMart interface for protozoa, plants, metazoans and fungi.
You can use the functions listEnsemblGenomes() and useEnsemblGenomes() in a similar way to the functions shown earlier. For example, first we can list the available Ensembl Genomes marts:
> listEnsemblGenomes()
biomart version
1 protists_mart Ensembl Protists Genes 51
2 protists_variations Ensembl Protists Variations 51
3 fungi_mart Ensembl Fungi Genes 51
4 fungi_variations Ensembl Fungi Variations 51
5 metazoa_mart Ensembl Metazoa Genes 51
6 metazoa_variations Ensembl Metazoa Variations 51
7 plants_mart Ensembl Plants Genes 51
8 plants_variations Ensembl Plants Variations 51
We can select the Ensembl Plants Database and search the dataset name of Arabidopsis.
> ensembl_plants <- useEnsemblGenomes(biomart = "plants_mart")
> searchDatasets(ensembl_plants, pattern = "Arabidopsis")
dataset description version
4 ahalleri_eg_gene Arabidopsis halleri genes (Ahal2.2) Ahal2.2
5 alyrata_eg_gene Arabidopsis lyrata genes (v.1.0) v.1.0
8 athaliana_eg_gene Arabidopsis thaliana genes (TAIR10) TAIR10
We can then use this information to create our Mart object, which will access the correct database and dataset.
> ensembl_arabidopsis <- useEnsemblGenomes(biomart = "plants_mart", dataset = "athaliana_eg_gene")
How to build a biomaRt query
Once we have selected the dataset from which to get the data, we need to create a query and send it to the Ensembl BioMart server. We use the getBM() function to do this.
The getBM() function has three parameters to be introduced: filter, value, and attribute.
Filters and values are used to define restrictions on queries. For example, if you want to limit the output to all genes on the human X chromosome, you can use the filter chromosome with value "X"_ name. The listFilters() function displays all available filters in the selected dataset.
> filters = listFilters(ensembl)
> filters[1:5,]
name description
1 chromosome_name Chromosome/scaffold name
2 start Start
3 end End
4 band_start Band Start
5 band_end Band End
Attributes define the data we are interested in retrieving. For example, maybe we want to retrieve gene symbols or chromosomal coordinates. The listAttributes() function displays all available attributes in the selected dataset.
> attributes = listAttributes(ensembl)
> attributes[1:5,]
name description page
1 ensembl_gene_id Gene stable ID feature_page
2 ensembl_gene_id_version Gene stable ID version feature_page
3 ensembl_transcript_id Transcript stable ID feature_page
4 ensembl_transcript_id_version Transcript stable ID version feature_page
5 ensembl_peptide_id Protein stable ID feature_page
The getBM() function is the main query function in biomaRt. It has four main arguments:
- attributes: is an attribute vector to retrieve (= output of the query).
- filters: is a filter vector used as input to the query.
- Values: the value vector of the filter. If multiple filters are used, the values parameter requires a list of values, where each position in the list corresponds to the position of the filter in the filter parameter (see the following example).
- Mart: an object of the mart class, created by the useEnsembl() function.
Note: for some common queries of Ensembl, you can use wrapper functions: getGene() and getSequence(). These functions call the getBM() function with hard coded filters and attribute names.
Now we have selected the biomaRt database and dataset, and understood the attributes, filters and filter values; We can build a biomaRt query. Let's make a simple query on the following question: we have a list of Affymetrix identifiers from u133plus2 platform, and we want to use Ensembl mapping to retrieve the corresponding EntrezGene identifier.
The u133plus2 platform will be the filter for this query. As the value of this filter, we use our Affymetrix identifier list. As the output (attribute) of the query, we want to retrieve the EntrezGene and u133plus2 identifiers, so we get the mapping results of these two identifiers. The exact names we must use to specify the attributes and filters can be retrieved using the listAttributes() and listFilters() functions, respectively. Now let's run the query:
> affyids <- c("202763_at","209310_s_at","207500_at")
> getBM(attributes = c('affy_hg_u133_plus_2', 'entrezgene_id'), filters = 'affy_hg_u133_plus_2', values = affyids, mart = ensembl)
affy_hg_u133_plus_2 entrezgene_id
1 202763_at 836
2 209310_s_at 837
3 207500_at 838
3.1 search for filters and attributes
The functions listAttributes() and listFilters() will return each available option of their respective types, which will produce long output and it will be difficult to find the value you are interested in. biomaRt also provides the functions searchAttributes() and searchFilters(), which will try to find any entries that match a specific term or pattern in a similar way to the searchDatasets() you saw earlier. You can use these functions to find available properties and filters that you might be interested in. The following example returns details of all the properties that contain the schema "hgnc".
> searchAttributes(mart = ensembl, pattern = "hgnc")
name description page
64 hgnc_id HGNC ID feature_page
65 hgnc_symbol HGNC symbol feature_page
96 hgnc_trans_name Transcript name ID feature_page
For advanced use, note that the pattern parameters are regular expressions. This means that you can create more complex queries as needed. Imagine, for example, that we have the string ENST00000577249.1, which we know is some kind of Ensembl ID, but we're not sure what the appropriate filter term is. The example shown next uses a pattern that finds all filters that contain the terms "ensembl" and "id". This allows us to reduce the filter list to one that is only applicable to our example.
> searchFilters(mart = ensembl, pattern = "ensembl.*id")
name description
57 ensembl_gene_id Gene stable ID(s) [e.g. ENSG00000000003]
58 ensembl_gene_id_version Gene stable ID(s) with version [e.g. ENSG00000000003.15]
59 ensembl_transcript_id Transcript stable ID(s) [e.g. ENST00000000233]
60 ensembl_transcript_id_version Transcript stable ID(s) with version [e.g. ENST00000000233.10]
61 ensembl_peptide_id Protein stable ID(s) [e.g. ENSP00000000233]
62 ensembl_peptide_id_version Protein stable ID(s) with version [e.g. ENSP00000000233.5]
63 ensembl_exon_id Exon ID(s) [e.g. ENSE00000000003]
Thus, we can compare ENST00000577249.1 with the example given in the Description column, and we can see that it is a Transcript ID with version. Therefore, the appropriate filter value to use with it is ensembl_transcript_id_version.
3.2 use predefined filter values
Many filters have a predefined list of values that are known in their associated dataset. A common example is the name of a chromosome when Ensembl searches a dataset. In this online interface of BioMart, these available options are displayed as a list, as shown in Figure 1.
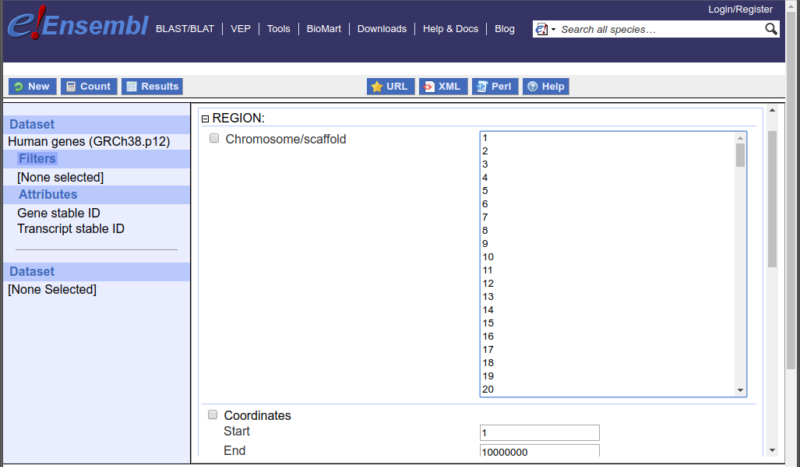
Figure 1: the options available in the chromosome / Scaffold field are limited to a predetermined list based on the values in this dataset
You can use the function listFilterOptions() to list it in the R session, passing it the name of a Mart object and filter. For example, to list possible chromosome names, you can run the following command:
> listFilterOptions(mart = ensembl, filter = "chromosome_name") [1] "1" "2" "3" [4] "4" "5" "6" [7] "7" "8" "9" [10] "10" "11" "12" [13] "13" "14" "15" [16] "16" "17" "18" [19] "19" "20" "21"
You can also search the list of available values through searchFilterOptions(). In the following two examples, the first returns all chromosome names starting with "GL", while the second looks for any phenotypic description containing the string "Crohn".
> searchFilterOptions(mart = ensembl, filter = "chromosome_name", pattern = "^GL") [1] "GL000009.2" "GL000194.1" "GL000195.1" "GL000205.2" "GL000213.1" "GL000216.2" "GL000218.1" "GL000219.1" "GL000220.1" "GL000225.1" > searchFilterOptions(mart = ensembl, filter = "phenotype_description", pattern = "Crohn") [1] "INFLAMMATORY BOWEL DISEASE CROHN DISEASE 1" "INFLAMMATORY BOWEL DISEASE CROHN DISEASE 10" "INFLAMMATORY BOWEL DISEASE CROHN DISEASE 19" [4] "INFLAMMATORY BOWEL DISEASE CROHN DISEASE 30" "NON RARE IN EUROPE: Crohn disease"
3.3 find out more about filters
3.3.1 filter type
Boolean filter requires a value of TRUE or FALSE in biomaRt. A setting of TRUE will include all information that meets the filter requirements. Setting FALSE will exclude information that meets the filter requirements and return all values that do not meet the filter requirements. For most filters, their names indicate whether the type is Boolean, and they usually start with "with". However, this is not a rule. To ensure that your type is correct, you can use the function filterType() to investigate the type of filter you want to use.
> filterType("with_affy_hg_u133_plus_2", ensembl)
[1] "boolean_list"
3.4 property page
For large BioMart databases such as Ensembl, the listAttributes() function may display a very large number of attributes. In the BioMart database, attributes are placed on the page, such as sequences, features, Ensembl's homes. You can use the attributePages() function to get an overview of the attribute pages that exist in each BioMart dataset.
> pages = attributePages(ensembl) > pages [1] "feature_page" "structure" "homologs" "snp" "snp_somatic" "sequences"
To show us a small list of attributes belonging to a particular page, we can now specify it in the listAttributes() function.
> head(listAttributes(ensembl, page="feature_page"))
name description page
1 ensembl_gene_id Gene stable ID feature_page
2 ensembl_gene_id_version Gene stable ID version feature_page
3 ensembl_transcript_id Transcript stable ID feature_page
4 ensembl_transcript_id_version Transcript stable ID version feature_page
5 ensembl_peptide_id Protein stable ID feature_page
6 ensembl_peptide_id_version Protein stable ID version feature_page
We now have a short list of attributes related to the region where the gene is located.
3.5 using select()
To provide a more consistent interface for all annotations in the Bioconductor, select(), columns(), keytypes(), and keys() have been implemented to wrap some of the existing functions above. These methods are called the same way they are used in other parts of the project, except that they do not take the AnnotationDb derived class, but take the Mart derived class as their first parameter. Otherwise, the usage should be basically the same. You still use columns() to discover what can be extracted from the Mart and keytypes() to discover what can be used as the key for select().
> mart <- useEnsembl(dataset = "hsapiens_gene_ensembl", biomart='ensembl') > head(keytypes(mart),n=3) [1] "affy_hc_g110" "affy_hg_focus" "affy_hg_u133_plus_2" > head(columns(mart), n = 3) [1] "3_utr_end" "3_utr_end" "3_utr_start"
You can still use keys() to extract potential keys for a particular key type.
> k = keys(mart, keytype="chromosome_name") > head(k, n=3) [1] "1" "2" "3"
With keys(), you can even take advantage of the additional parameters available to other key methods
> k = keys(mart, keytype="chromosome_name", pattern="LRG") > head(k, n=3) character(0)
Unfortunately, the keys() method is not applicable to all key types because they are not all supported.
But you can still use select() here to extract data columns that match a particular keyset (this is basically a wrapper for getBM()).
> affy=c("202763_at","209310_s_at","207500_at")
> select(mart, keys=affy, columns=c('affy_hg_u133_plus_2','entrezgene_id'), keytype='affy_hg_u133_plus_2')
affy_hg_u133_plus_2 entrezgene_id
1 202763_at 836
2 209310_s_at 837
3 207500_at 838
So why do we do this when we already have a function like getBM()? There are two reasons: 1) for those who are familiar with select and its auxiliary methods, they can now continue to use biomaRt to make the same type of calls they are already familiar with; 2) Because select methods are implemented in many places elsewhere, the fact that these methods are shared allows more convenient programmatic access to all these resources. An example package that takes advantage of this is the OrganismDbi package. Multiple packages can be accessed as one resource.
Result cache
To save time and computational resources, biomaRt will try to identify when you rerun the previously executed query. Each time you run a new query, the results are saved to the cache on your computer. If a query is recognized as having been run before instead of submitting the query to the server, the results are loaded from the cache.
You can use the biomartCacheInfo() function to get some information about the cache size and location:
> biomartCacheInfo() biomaRt cache - Location: C:\Users\MSI\AppData\Local/biomaRt/biomaRt/Cache - No. of files: 2 - Total size: 239 bytes
You can delete the cache using the command biomartCacheClear(). This will delete all cached files.
The default location of the cache is specific to your computer and operating system. If you want to use a specific location, you can use biomaRt_ Cache environment variable. You can set it outside R, or you can set it in R by calling sys.setenv (BIOMART_CACHE = "< / where / I / store / my / cache >"). The following code gives an example, we change the location to a temporary file, and then confirm that the location has changed.
> Sys.setenv(BIOMART_CACHE = tempdir()) > biomartCacheInfo() biomaRt cache - Location: C:\Users\MSI\AppData\Local\Temp\Rtmp02rPad - No. of files: 0 - Total size: 0 bytes
biomaRt auxiliary function
This section describes a set of biomaRt helper functions that can be used to export FASTA format sequences, retrieve the values of some filters, and explore available filters and attributes in a more systematic manner.
5.1 exportFASTA
You can use the exportFASTA() function to export the data.frames obtained by the getSequence() function to a FASTA file. You must use the file parameter to specify the data.frame and file name to export.
biomaRt query example
In the following sections, various sample queries are described. Each example is written as a task, and we must come up with a biomaRt solution to solve this problem.
6.1 annotate a set of Affymetrix identifiers with HUGO symbols and chromosome positions of corresponding genes
We have a list of Affymetrix hgu133plus2 identifiers. We want to retrieve HUGO gene symbols, chromosome names, start and end positions and bands of corresponding genes. The listAttributes() and listFilters() functions give us an overview of the available attributes and filters, and we look in these lists for the corresponding attribute and filter names we need. For this query, we need the following properties: hgnc_symbol,chromsome_name,start_position,end_position, band and Affy_ hg_ u133_ plus_ 2 (because we want these attributes in the output to provide a mapping with the original Affymetrix input identifier. There is a filter in this query, which is affy_hg_u133_plus_2 filter, because we use the list of Affymetrix identifiers as input. Put all these in getBM() and execute the query to give:
> affyids=c("202763_at","209310_s_at","207500_at")
> getBM(attributes = c('affy_hg_u133_plus_2', 'hgnc_symbol', 'chromosome_name', 'start_position', 'end_position', 'band'), filters = 'affy_hg_u133_plus_2', values = affyids, mart = ensembl)
affy_hg_u133_plus_2 hgnc_symbol chromosome_name start_position end_position band
1 202763_at CASP3 4 184627696 184649509 q35.1
2 209310_s_at CASP4 11 104942866 104969366 q22.3
3 207500_at CASP5 11 104994235 105023168 q22.3
6.2 annotate a set of EntrezGene identifiers with GO annotation
In this task, we start with a list of entrezgene identifiers. We want to retrieve GO identifiers related to biological processes associated with these entrezgene identifiers. We check the output of listAttributes() and listFilters() again to find the filters and attributes we need. Then we construct the following query:
> entrez=c("673","837")
> goids = getBM(attributes = c('entrezgene_id', 'go_id'), filters = 'entrezgene_id', values = entrez, mart = ensembl)
> head(goids)
entrezgene_id go_id
1 673 GO:0005524
2 673 GO:0007165
3 673 GO:0006468
4 673 GO:0035556
5 673 GO:0004672
6 673 GO:0043231
6.3 retrieve all HUGO gene symbols of genes located on chromosomes 17, 20 or Y and associated with specific GO terms
The GO terms we are interested in are: GO:0051330, GO:0000080, GO:0000114, GO:0000082. The key to executing this query is to understand getBM() The function enables you to use multiple filters at the same time. To do this, the filter parameter should be a vector with the filter name. These values should be a list, where the first element of the list corresponds to the first filter, the second list element corresponds to the second filter, and so on. The elements of this list are possible to contain the corresponding filters Vector of values.
> go=c("GO:0051330","GO:0000080","GO:0000114","GO:0000082")
> chrom=c(17,20,"Y")
> getBM(attributes= "hgnc_symbol",filters=c("go","chromosome_name"),values=list(go, chrom), mart=ensembl)
hgnc_symbol
1 E2F1
2 CRLF3
3 RPS6KB1
4 CDK3
6.4 annotate the identifier set using the INTERPRO protein domain identifier
In this example, we want to annotate the following two RefSeq identifiers with INTERPRO protein domain identifier and protein domain description: NM_005359 and NM_000546.
> refseqids = c("NM_005359","NM_000546")
> ipro = getBM(attributes=c("refseq_mrna","interpro","interpro_description"), filters="refseq_mrna", values=refseqids, mart=ensembl)
> ipro
refseq_mrna interpro interpro_description
1 NM_000546 IPR002117 p53 tumour suppressor family
2 NM_000546 IPR008967 p53-like transcription factor, DNA-binding
3 NM_000546 IPR010991 p53, tetramerisation domain
4 NM_000546 IPR011615 p53, DNA-binding domain
5 NM_000546 IPR012346 p53/RUNT-type transcription factor, DNA-binding domain superfamily
6 NM_000546 IPR013872 p53 transactivation domain
7 NM_000546 IPR036674 p53-like tetramerisation domain superfamily
8 NM_000546 IPR040926 Cellular tumor antigen p53, transactivation domain 2
9 NM_005359 IPR001132 SMAD domain, Dwarfin-type
10 NM_005359 IPR003619 MAD homology 1, Dwarfin-type
11 NM_005359 IPR008984 SMAD/FHA domain superfamily
12 NM_005359 IPR013019 MAD homology, MH1
13 NM_005359 IPR013790 Dwarfin
14 NM_005359 IPR017855 SMAD-like domain superfamily
15 NM_005359 IPR036578 SMAD MH1 domain superfamily
6.5 select all Affymetrix identifiers on hgu133plus2 chip and Ensembl gene identifiers of genes located between 1100000 and 1250000 base pairs on chromosome 16.
In this example, we will use multiple filters again: chromosome_name, start and end, because we filter these three conditions. Note that when chromosome name, start position and end position are used together as filters, the BioMart network service interprets them as returning all the contents of a given chromosome between a given start and end position.
> getBM(attributes = c('affy_hg_u133_plus_2','ensembl_gene_id'), filters = c('chromosome_name','start','end'),values = list(16,1100000,1250000), mart = ensembl)
affy_hg_u133_plus_2 ensembl_gene_id
1 ENSG00000260702
2 215502_at ENSG00000260532
3 ENSG00000273551
4 205845_at ENSG00000196557
5 ENSG00000196557
6 ENSG00000260403
7 ENSG00000259910
8 ENSG00000261294
9 220339_s_at ENSG00000116176
10 ENSG00000277010
11 205683_x_at ENSG00000197253
12 207134_x_at ENSG00000197253
13 217023_x_at ENSG00000197253
14 210084_x_at ENSG00000197253
15 215382_x_at ENSG00000197253
16 216474_x_at ENSG00000197253
17 205683_x_at ENSG00000172236
18 207134_x_at ENSG00000172236
19 217023_x_at ENSG00000172236
20 210084_x_at ENSG00000172236
21 215382_x_at ENSG00000172236
22 216474_x_at ENSG00000172236
6.6 retrieve all EntrezGene identifiers and HUGO gene symbols of genes with "MAP kinase activity" GO term.
The GO identifier for MAP kinase activity is GO:0004707. In our query, we will use go_id as the filter and entrezgene_id and hgnc_symbol as the attributes. This is the query:
> getBM(attributes = c('entrezgene_id','hgnc_symbol'), filters = 'go', values = 'GO:0004707', mart = ensembl)
entrezgene_id hgnc_symbol
1 225689 MAPK15
2 5596 MAPK4
3 5599 MAPK8
4 5594 MAPK1
5 51701 NLK
6 6300 MAPK12
7 5600 MAPK11
8 5595 MAPK3
9 5602 MAPK10
10 1432 MAPK14
11 5603 MAPK13
12 5597 MAPK6
13 5598 MAPK7
14 5601 MAPK9
6.7 given a set of EntrezGene identifiers, 100 bp upstream promoter sequence was retrieved
All sequence related queries to Ensembl can be obtained through the getSequence() wrapper function. getBM() can also be used directly to retrieve sequences, but this may become complex, so getSequence() is provided as a general function suitable for most cases.
You can use the getSequence() function to retrieve the sequence from the chromosome coordinates or identifiers.
The chromosome name can be specified using the chromosome parameter. The start and end parameters are used to specify the start and end positions on the chromosome. The returned sequence type can be specified by the seqType parameter, which takes the following values:
- cdna
- Peptide of protein sequence
- 3utr represents the 3 'UTR sequence
- 5utr represents 5 'UTR sequence
- gene_exon indicates only for exon sequences
- transcript_exon indicates an exon sequence that is specific to the transcript only
- ranscript_exon_intron indicates that a complete non spliced transcript is given, i.e. exon + intron
- gene_exon_intron gives the exon + intron of the gene
- Coding only gives the coding sequence
- Coding_script_frank gives the flanking region of transcripts including UTR, which must be accompanied by a given upstream or downstream attribute value
- Coding_gene_frank gives the flanking region of genes including UTR, which must be accompanied by a given upstream or downstream attribute value
- Script_frank gives the flanking region of transcripts excluding UTR, which must be accompanied by the given value of upstream or downstream attributes
- Gene_frank gives the flanking region of genes that do not include UTR, which must be accompanied by a given upstream or downstream attribute value
This task requires us to retrieve 100 bp upstream promoter sequence from a set of EntrezGene identifiers. The type parameter in getsequence () can be considered a filter in this query and uses the same input name given by listFilters(). In our query, we use entrezgene_id as a type parameter. Next, we must specify the type of sequence we want to retrieve. Here, we are interested in the sequence of the promoter region, starting next to the coding of the gene. Set seqType to coding_ gene_ Frank will provide what we need. The upstream parameter is used to specify how many bp upstream sequences we want to retrieve. Here, we will retrieve a fairly short 100 bp sequence. All of these are given in getSequence():
> entrez=c("673","7157","837")
> getSequence(id = entrez,
+ type="entrezgene_id",
+ seqType="coding_gene_flank",
+ upstream=100,
+ mart=ensembl)
## coding_gene_flank
## 1 TCCTTCTCTGCAGGCCCAGGTGACCCAGGGTTGGAAGTGTCTCATGCTGGATCCCCACTTTTCCTCTTGCAGCAGCCAGACTGCCTTCCGGGTCACTGCC
## 2 CCTCCGCCTCCGCCTCCGCCTCCGCCTCCCCCAGCTCTCCGCCTCCCTTCCCCCTCCCCGCCCGACAGCGGCCGCTCGGGCCCCGGCTCTCGGTTATAAG
## 3 CACGTTTCCGCCCTTTGCAATAAGGAAATACATAGTTTACTTTCATTTTTGACTCTGAGGCTCTTTCCAACGCTGTAAAAAAGGACAGAGGCTGTTCCCT
## entrezgene_id
## 1 7157
## 2 673
## 3 837
Error in .processResults(postRes, mart = mart, hostURLsep = sep, fullXmlQuery = fullXmlQuery, :
Query ERROR: caught BioMart::Exception::Usage: Filter upstream_flank NOT FOUND
It should also be noted that although we search for genes based on NCBI gene ID, Ensembl BioMart does not allow direct return of some ID types (including NCBI ID). In order to try and adapt to this biomaRt, try to map the query ID to the Ensembl gene ID internally before finding the sequence information. If no such mapping exists (or at least not found in Ensembl), no sequence is returned for the affected ID.
6.8 search all 5'UTR sequences of all genes located between chromosome 3 185514033 and 185535839
As described in the previous task, getSequence() can also use chromosome coordinates to retrieve the sequences of all genes located in a given region. We must also specify the identifier type to retrieve with the sequence. Here we select NCBI Gene ID:
> utr5 = getSequence(chromosome=3, start=185514033, end=185535839, type="entrezgene_id", seqType="5utr", mart=ensembl)
> utr5
5utr entrezgene_id
1 TGAGCAAAATCCCACAGTGGAAACTCTTAAGCCTCTGCGAAGTAAATCATTCTTGTGAATGTGACACACGATCTCTCCAGTTTCCAT 200879
2 ACCACACCTCTGAGTCGTCTGAGCTCACTGTGAGCAAAATCCCACAGTGGAAACTCTTAAGCCTCTGCGAAGTAAATCATTCTTGTGAATGTGACACACGATCTCTCCAGTTTCCAT 200879
3 ATTCTTGTGAATGTGACACACGATCTCTCCAGTTTCCAT 200879
4 Sequence unavailable 200879
6.9 retrieve the protein sequence of the given EntrezGene identifier list
In this task, the type parameter specifies the type of identifier we use. For an overview of other valid identifier types, refer to the listFilters() function.
> protein = getSequence(id=c(100, 5728), type="entrezgene_id", seqType="peptide", mart=ensembl)
> protein
peptide
1 ALLFHKMMFETIPMFSGGTCNPQFVVCQLKVKIYSSNSGPTRREDKFMYFEFPQPLPVCGDIKVEFFHKQNKMLKKDKMFHFWVNTFFIPGPEETSEKVENGSLCDQEIDSICSIERADNDKEYLVLTLTKNDLDKANKDKANRYFSPNFKVS*
2 MTAIIKEIVSRNKRRYQEDGFDLDLTYIYPNIIAMGFPAERLEGVYRNNIDDVVRFLDSKHKNHYKIYNLCAERHYDTAKFNCRVAQYPFEDHNPPQLELIKPFCEDLDQWLSEDDNHVAAIHCKAGKGRTGVMICAYLLHRGKFLKAQEALDFYGEVRTRDKKGVTIPSQRRYVYYYSYLLKNHLDYRPVALLFHKMMFETIPMFSGGTCNPQFVVCQLKVKIYSSNSGPTRREDKFMYFEFPQPLPVCGDIKVEFFHKQNKMLKKDKMFHFWVNTFFIPGPEETSEKVENGSLCDQEIDSICSIERADNDKEYLVLTLTKNDLDKANKDKANRYFSPNFKVKLYFTKTVEEPSNPEASSSTSVTPDVSDNEPDHYRYSDTTDSDPENEPFDEDQHTQITKV*
3 Sequence unavailable
4 Sequence unavailable
5 MAQTPAFDKPKVELHVHLDGSIKPETILYYGRRRGIALPANTAEGLLNVIGMDKPLTLPDFLAKFDYYMPAIAGCREAIKRIAYEFVEMKAKEGVVYVEVRYSPHLLANSKVEPIPWNQAEGDLTPDEVVALVGQGLQEGERDFGVKARSILCCMRHQPNWSPKVVELCKKYQQQTVVAIDLAGDETIPGSSLLPGHVQAYQEAVKSGIHRTVHAGEVGSAEVVKEAVDILKTERLGHGYHTLEDQALYNRLRQENMHFEAQK*
6 MAQTPAFDKPKVELHVHLDGSIKPETILYYGRRRGIALPANTAEGLLNVIGMDKPLTLPDFLAKFDYYMPAIARL*
7 MAQTPAFDKPKVELHVHLDGSIKPETILYYGRRRGIALPANTAEGLLNVIGMDKPLTLPDFLAKFDYYMPAIAGCREAIKRIAYEFVEMKAKEGVVYVEVRYSPHLLANSKVEPIPWNQAEGDLTPDEVVALVGQGLQEGERDFGVKARSILCCMRHQPNWSPKVVELCKKYQQQTVVAIDLAGDETIPGSSLLPGHVQAYQEAVKSGIHRTVHAGEVGSAEVVKEAVDILKTERLGHGYHTLEDQALYNRLRQENMHFEICPWSSYLTGAWKPDTEHAVIRLKNDQANYSLNTDDPLIFKSTLDTDYQMTKRDMGFTEEEFKRLNINAAKSSFLPEDEKRELLDLLYKAYGMPPSASAGQNL*
8 MAQTPAFDKPKVELHVHLDGSIKPETILYYGRRRGIALPANTAEGLLNVIGMDKPLTLPDFLAKFDYYMPAIAGCREAIKRIAYEFVEMKAKEGVVYVEVRYSPHLLANSKVEPIPWNQAEGDLTPDEVVALVGQGLQEGERDFGVKARSILCCMRHQPNWSPKVVELCKKYQQQTVVAIDLAGDETIPGSSLLPGHVQAYQAVDILKTERLGHGYHTLEDQALYNRLRQENMHFEICPWSSYLTGAWKPDTEHAVIRLKNDQANYSLNTDDPLIFKSTLDTDYQMTKRDMGFTEEEFKRLNINAAKSSFLPEDEKRELLDLLYKAYGMPPSASAGQNL*
9 LERGGEAAAAAAAAAAAPGRGSESPVTISRAGNAGELVSPLLLPPTRRRRRRHIQGPGPVLNLPSAAAAPPVARAPEAAGGGSRSEDYSSSPHSAAAAARPLAAEEKQAQSLQPSSSRRSSHYPAAVQSQAAAERGASATAKSRAISILQKKPRHQQLLPSLSSFFFSHRLPDMTAIIKEIVSRNKRRYQEDGFDLDLTYIYPNIIAMGFPAERLEGVYRNNIDDVVRFLDSKHKNHYKIYNLCAERHYDTAKFNCRVAQYPFEDHNPPQLELIKPFCEDLDQWLSEDDNHVAAIHCKAGKGRTGVMICAYLLHRGKFLKAQEALDFYGEVRTRDKKGVTIPSQRRYVYYYSYLLKNHLDYRPVALLFHKMMFETIPMFSGGTCNPQFVVCQLKVKIYSSNSGPTRREDKFMYFEFPQPLPVCGDIKVEFFHKQNKMLKKDKMFHFWVNTFFIPGPEETSEKVENGSLCDQEIDSICSIERADNDKEYLVLTLTKNDLDKANKDKANRYFSPNFKVKLYFTKTVEEPSNPEASSSTSVTPDVSDNEPDHYRYSDTTDSDPENEPFDEDQHTQITKV*
entrezgene_id
1 5728
2 5728
3 5728
4 100
5 100
6 100
7 100
8 100
9 5728
6.10 search for known SNP s located between 148350 and 148420 on human chromosome 8
For this example, we must first connect to a different BioMart database, snp.
> snpmart = useEnsembl(biomart = "snp", dataset="hsapiens_snp")
The listAttributes() and listFilters() functions give us an overview of the available attributes and filters.
From these we need: refsnp_id, allele, chrom_start and chrome_ Strand as attribute; As a filter, we will use: chrome_ start,chrom_end and chr_name.
Put the attribute and filter we selected into getBM() to give:
> getBM(attributes = c('refsnp_id','allele','chrom_start','chrom_strand'), filters = c('chr_name','start','end'), values = list(8, 148350, 148420), mart = snpmart)
refsnp_id allele chrom_start chrom_strand
1 rs1450830176 G/C 148350 1
2 rs1360310185 C/T 148352 1
3 rs1434776028 A/T 148353 1
4 rs1413161474 C/T 148356 1
5 rs1410590268 A/G 148365 1
6 rs1193735780 T/A/C 148368 1
7 rs1409139861 C/T 148371 1
8 rs868546642 A/G 148372 1
9 rs547420070 A/C 148373 1
10 rs1236874674 C/T 148375 1
11 rs1207902742 C/T 148376 1
12 rs1437239557 T/C 148377 1
13 rs1160135941 T/G 148379 1
14 rs1229249227 A/T 148380 1
15 rs1584865972 C/G 148381 1
16 rs1328678285 C/G 148390 1
17 rs77274555 G/A/C 148391 1
18 rs567299969 T/A/C 148394 1
19 rs1457776094 A/C/G 148395 1
20 rs1292078334 C/T 148404 1
21 rs1456392146 A/T 148405 1
22 rs368076569 G/A 148407 1
23 rs1166604298 A/G 148408 1
24 rs1393545673 A/G/T 148410 1
25 rs1180076939 A/T 148413 1
26 rs1476313471 A/G 148414 1
27 rs1248424402 T/C 148417 1
28 rs1207939741 A/T 148419 1
6.11 given the human gene TP53, search the human chromosome position of the gene, and search the chromosome position and RefSeq id of its homologous gene in mice.
The getLDS() (get linked dataset) function provides the function of linking two BioMart datasets to each other and building queries on the two datasets. In Ensembl, linking two data sets is transformed into retrieving homology data across species.
The usage of getLDS is very similar to getBM(). Linked datasets are provided by separate Mart objects, and filters and properties must be specified for linked datasets. Filters can be applied to both datasets or one of them. Use the listFilters and listAttributes functions on two Mart objects to find the filters and attributes for each dataset (species in Ensembl). Attributes and filters for linked datasets can be specified using the attributesL and filtersL parameters. Enter all this information into getLDS() to give:
> human <- useEnsembl("ensembl", dataset = "hsapiens_gene_ensembl")
> mouse <- useEnsembl("ensembl", dataset = "mmusculus_gene_ensembl")
> getLDS(attributes = c("hgnc_symbol","chromosome_name", "start_position"), filters = "hgnc_symbol", values = "TP53", mart = human, attributesL = c("refseq_mrna","chromosome_name","start_position"), martL = mouse)
HGNC.symbol Chromosome.scaffold.name Gene.start..bp. RefSeq.mRNA.ID Chromosome.scaffold.name.1 Gene.start..bp..1
1 TP53 17 7661779 11 69471185
2 TP53 17 7661779 NM_001127233 11 69471185
3 TP53 17 7661779 NM_011640 11 69471185
Connection troubleshooting
It is not uncommon to encounter connection problems when trying to access online resources such as Ensembl BioMart. In this section, we list the error messages reported by the user and the suggested code to fix the problem. If the proposed solution does not work, or if you have new errors not listed here, please report them on the Bioconductor support site.
7.1 biomaRt specific solutions
If you use biomaRt directly, make sure you use useEnsembl() to create a Mart object, not useMart(). useEnsembl() knows some of the specific connection details required to connect to Ensembl, and it can be used to solve any connection problems without any further action from you.
7.2 global connection settings
If you cannot modify the biomaRt code (for example, if you use another package to call biomaRt as part of its functionality), you can still modify the connection settings of the R session. Here are some reported error messages and code known to resolve them. You only need to execute this code once in an R session, but the settings do not remain the same between R sessions.
7.2.1 Error: "SSL certificate problem"
Error message
Error in curl::curl_fetch_memory(url, handle = handle) : SSL certificate problem: unable to get local issuer certificate
Fix
httr::set_config(httr::config(ssl_verifypeer = FALSE))
7.2.1 Error: "sslv3 alert handshake failure"
Error message
Error in curl::curl_fetch_memory(url, handle = handle) : error:14094410:SSL routines:ssl3_read_bytes:sslv3 alert handshake failure
Fix
If you are running Ubuntu 20.04 or later, the following command should solve the problem.
httr::set_config(httr::config(ssl_cipher_list = "DEFAULT@SECLEVEL=1"))
If you encounter this error on Fedora 33, the above code does not seem to work. At present, the only solution we have found is to change the security settings at the system level. Check out fedoraproject.org for more information and GitHub for troubleshooting discussions. You can apply this change by running the following command in a terminal other than R, but consider whether this is what you want to change. You can also consider reminding Ensembl of this problem.
update-crypto-policies --set LEGACY
Session Info
> sessionInfo() R version 4.1.1 (2021-08-10) Platform: x86_64-w64-mingw32/x64 (64-bit) Running under: Windows 10 x64 (build 19042) Matrix products: default locale: [1] LC_COLLATE=Chinese (Simplified)_China.936 LC_CTYPE=Chinese (Simplified)_China.936 LC_MONETARY=Chinese (Simplified)_China.936 [4] LC_NUMERIC=C LC_TIME=Chinese (Simplified)_China.936 attached base packages: [1] stats graphics grDevices utils datasets methods base other attached packages: [1] biomaRt_2.48.3 loaded via a namespace (and not attached): [1] KEGGREST_1.32.0 progress_1.2.2 tidyselect_1.1.1 purrr_0.3.4 vctrs_0.3.8 generics_0.1.0 stats4_4.1.1 [8] BiocFileCache_2.0.0 utf8_1.2.2 blob_1.2.2 XML_3.99-0.8 rlang_0.4.11 pillar_1.6.3 withr_2.4.2 [15] glue_1.4.2 DBI_1.1.1 rappdirs_0.3.3 BiocGenerics_0.38.0 bit64_4.0.5 dbplyr_2.1.1 GenomeInfoDbData_1.2.6 [22] lifecycle_1.0.1 stringr_1.4.0 zlibbioc_1.38.0 Biostrings_2.60.2 memoise_2.0.0 Biobase_2.52.0 IRanges_2.26.0 [29] fastmap_1.1.0 GenomeInfoDb_1.28.4 parallel_4.1.1 curl_4.3.2 AnnotationDbi_1.54.1 fansi_0.5.0 Rcpp_1.0.7 [36] filelock_1.0.2 cachem_1.0.6 S4Vectors_0.30.0 XVector_0.32.0 bit_4.0.4 hms_1.1.1 png_0.1-7 [43] digest_0.6.27 stringi_1.7.4 dplyr_1.0.7 tools_4.1.1 bitops_1.0-7 magrittr_2.0.1 RCurl_1.98-1.5 [50] RSQLite_2.2.8 tibble_3.1.4 crayon_1.4.1 pkgconfig_2.0.3 ellipsis_0.3.2 xml2_1.3.2 prettyunits_1.1.1 [57] assertthat_0.2.1 httr_1.4.2 rstudioapi_0.13 R6_2.5.1 compiler_4.1.1